RFdiffusion2 scaffolded all 41 of 41 enzyme active sites on an in-silico benchmark where the prior best deep-learning method managed 16 of 41.1 Read that again. Atom-level active-site scaffolding, one of the hardest problems in computational enzyme design, went from a 39 percent hit rate to a clean sweep in a single model generation. The authors then took it to the bench, found active catalysts for three distinct catalytic sites while screening fewer than 96 sequences each, and reported a best design with a kcat/KM of roughly 53,000 M^-1 s^-1, confirmed by X-ray crystallography.1
That single result captures both halves of where protein design sits in 2026. The generative machinery has made backbone and active-site generation almost routine on paper. And the field is still doing the slow, honest work of finding out how much of that paper performance survives contact with a pipette.
It helps to keep two ledgers while reading this literature. The first ledger is capability: what a model can demonstrably do in silico, measured by designability, self-consistency RMSD, sequence recovery, novelty, diversity. The second ledger is validity: what survives contact with an actual wet lab. Designs that express, fold, bind, and catalyze. The two ledgers diverge constantly, because the first is cheap to produce and easy to game, and the second costs money and months and is hard to fake. A benchmark number is a hypothesis about real-world performance, not a measurement of it.
This is a survey of the 14 models in the protein design and generation slice of the rewire.it Foundation Models in Biology catalog, the smallest of its six categories, sitting alongside structure prediction, protein language models, antibody-specific models, DNA models, and RNA models. The catalog has 14 entries, shown as 14 rows in the master table below, though they cover 16 named tools: two rows bundle a model with its variant (FrameDiff with FrameFlow, Genie with Genie 2), while the three RFdiffusion variants each get their own row despite being one family. I am going to organize the survey around the two ledgers, then walk the dominant two-stage workflow, the method families, the licensing split, and finally a decision tree you can act on. For an engineer, those last items, workflow and license and access, decide what you can actually deploy.
The two ledgers, stated plainly
A self-consistency designability score works like this. You generate a backbone. You design a sequence for it with ProteinMPNN. You fold that sequence back with ESMFold or AlphaFold2. You measure how far the prediction landed from your target backbone. If the self-consistency RMSD (scRMSD) is under 2 Angstrom and pLDDT is over 70, the design counts as "designable."2 SALAD uses exactly this definition: scRMSD under 2.0 Angstrom and ESMFold pLDDT over 70 over 8 ProteinMPNN sequences.2 Genie 2 uses the scTM variant, self-consistency TM over 0.5.3
The trouble is that every part of that loop is a neural network with shared inductive biases. ProteinMPNN and ESMFold both learned from the PDB. A backbone that fools an inverse-folding model into producing a sequence that an in-silico folder likes is a backbone that satisfies the consensus of two models trained on overlapping data. It has passed a conversation between two neural networks. It has not expressed, folded, or functioned. It is not yet a protein.
The validity ledger is the measurement. Here the units change: binder hit rate, measured affinity, expression yield, catalytic efficiency, X-ray confirmation. These cost reagents and time, so the papers that report them tend to report fewer designs and to be more honest about failure.
The diagram below sketches the standard pipeline so we can talk about where each ledger gets recorded.
 The dominant two-stage design pattern and where the two ledgers get recorded. Backbone generators feed inverse-folding sequence designers; some models co-generate or skip a stage. Sources: RFdiffusion,4 Genie 2,3 SALAD,2 FrameDiff/FrameFlow,56 ProteinMPNN,7 LigandMPNN,8 ESM-IF1,9 Chroma,10 EvoDiff,11 ProGen3,12 BindCraft,13 Chai-2.14
The dominant two-stage design pattern and where the two ledgers get recorded. Backbone generators feed inverse-folding sequence designers; some models co-generate or skip a stage. Sources: RFdiffusion,4 Genie 2,3 SALAD,2 FrameDiff/FrameFlow,56 ProteinMPNN,7 LigandMPNN,8 ESM-IF1,9 Chroma,10 EvoDiff,11 ProGen3,12 BindCraft,13 Chai-2.14
Keep that split in mind as we go through the models.
The master table
Here is the whole field in one view. Dates are release of the original version; the method and license columns decide most real choices. Bold names get fuller treatment below.
| Model | Developer | Released | Params | Method type | License (weights) |
|---|---|---|---|---|---|
| BindCraft | LPDI, EPFL + MIT | 2024.0913 | none trained (uses AF2 + ProteinMPNN)13 | AF2-hallucination pipeline13 | MIT, PyRosetta caveat15 |
| Chai-2 | Chai Discovery | 2025.0714 | undisclosed14 | all-atom generative + folding14 | closed/proprietary14 |
| Chroma | Generate Biomedicines | 2022.1210 | undisclosed10 | diffusion (backbone+seq)10 | non-commercial weights10 |
| ESM-IF1 | Meta FAIR | 20229 | 142M9 | inverse folding (GVPTransformer)9 | MIT9 |
| EvoDiff | Microsoft Research | 2023.0911 | 38M / 640M11 | discrete sequence diffusion11 | MIT11 |
| FrameDiff / FrameFlow | Yim et al., MIT CSAIL | 2023.025 | ~17.4M5 | SE(3) diffusion / flow matching56 | MIT5 |
| Genie / Genie 2 | AlQuraishi Lab, Columbia | 202316 | undisclosed16 | SE(3) diffusion (backbone)16 | Apache-2.03 |
| LigandMPNN | Baker Lab / IPD | 2023.128 | undisclosed (small)8 | inverse folding + ligand context8 | MIT8 |
| ProGen3 | Profluent | 2025.0412 | 112M to 46B; open ≤3B12 | autoregressive seq LM (MoE)12 | CC BY-NC-SA weights12 |
| ProteinMPNN | Baker Lab / IPD | 20227 | ~1.7M7 | inverse folding (MPNN)7 | MIT7 |
| RFdiffusion | Baker Lab / IPD | 20234 | undisclosed4 | diffusion on RoseTTAFold4 | BSD4 |
| RFdiffusion All-Atom | Baker Lab / IPD | 2023.1017 | undisclosed (~1GB)17 | all-atom diffusion (RFAA)17 | BSD17 |
| RFdiffusion2 | Baker Lab / IPD | 2025.041 | undisclosed1 | atomized diffusion1 | BSD-3-Clause1 |
| SALAD | Jendrusch & Korbel, EMBL | 2025.012 | ~8M2 | sparse diffusion2 | Apache-2.0 / CC-BY2 |
Two patterns jump out immediately. First, the models are small. Most of the structure-based generators are in the single-digit to low-tens of millions of parameters. ProteinMPNN is about 1.7 million.7 FrameDiff is about 17.4 million.5 SALAD is about 8 million.2 That smallest one, ProteinMPNN, is about one-fifth the size of ESM-2's smallest 8-million-parameter checkpoint, and roughly four-thousandths of a 460-million-parameter language model. Compare all of that to the one true heavyweight here, ProGen3, which spans 112 million to 46 billion parameters.12 Second, with three exceptions the weights are open and most are permissively licensed. We will return to both points.
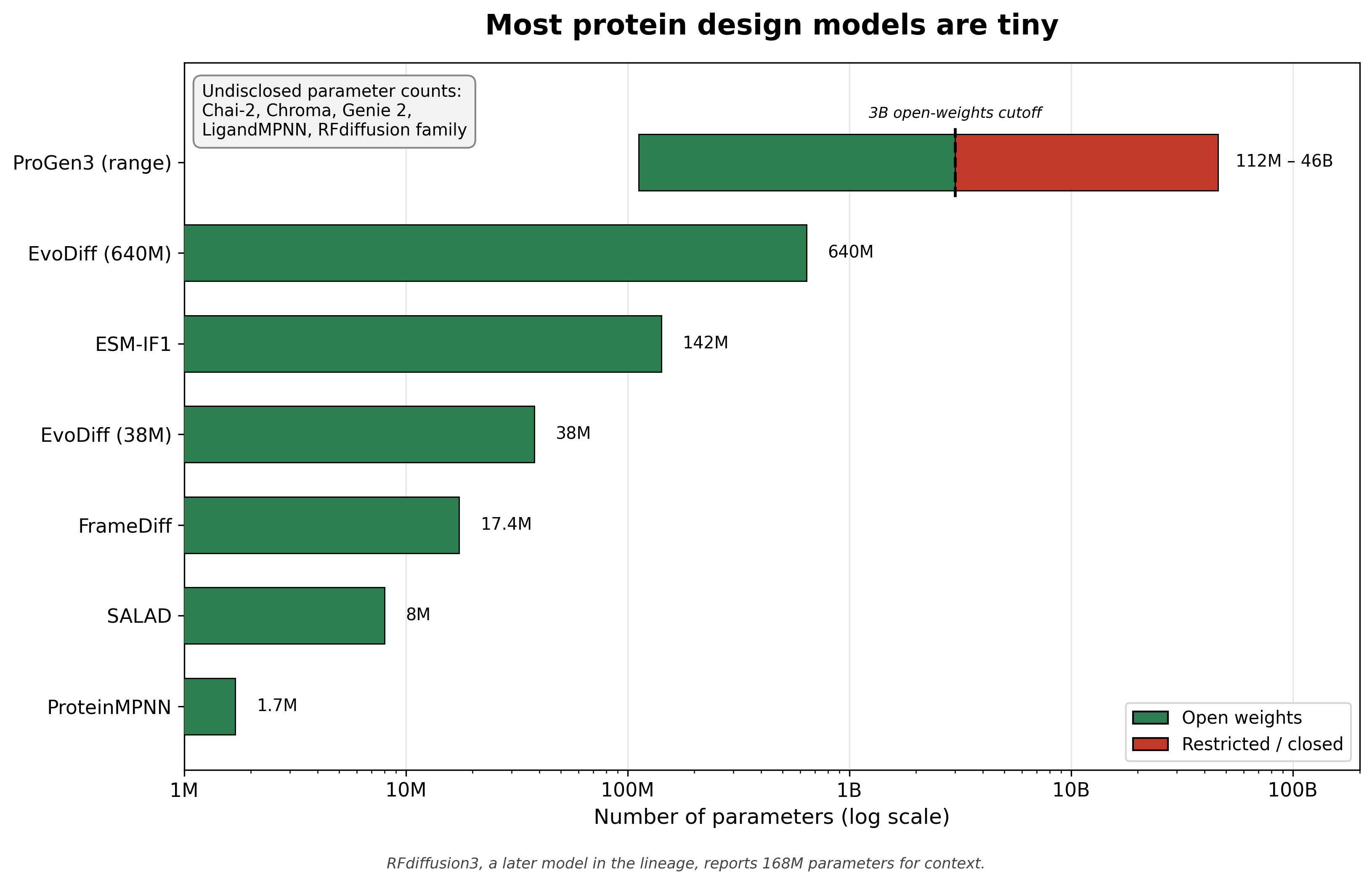 Parameter counts for the models that disclose them, log scale. ProteinMPNN ~1.7M,7 SALAD ~8M,2 FrameDiff ~17.4M,5 EvoDiff 38M and 640M,11 ESM-IF1 142M,9 ProGen3 spanning 112M to 46B with open weights capped at 3B.12 RFdiffusion,4 LigandMPNN,8 Genie 2,3 Chroma,10 and Chai-214 do not disclose parameter counts; for context, the later RFdiffusion3 reports 168M.18
Parameter counts for the models that disclose them, log scale. ProteinMPNN ~1.7M,7 SALAD ~8M,2 FrameDiff ~17.4M,5 EvoDiff 38M and 640M,11 ESM-IF1 142M,9 ProGen3 spanning 112M to 46B with open weights capped at 3B.12 RFdiffusion,4 LigandMPNN,8 Genie 2,3 Chroma,10 and Chai-214 do not disclose parameter counts; for context, the later RFdiffusion3 reports 168M.18
The capability ledger: what the benchmarks say
Group the field by method type and the capability story is fairly clean.
Inverse folding models take a fixed backbone and output a sequence. ProteinMPNN is the workhorse: a 3-layer-encoder, 3-layer-decoder message-passing network at about 1.7M parameters, trained on PDB assemblies clustered at 30 percent identity into 25,361 clusters, reporting 52.4 percent native sequence recovery versus 32.9 percent for Rosetta.7 ESM-IF1 is the other classic, a 142M GVPTransformer (4 GVP-GNN layers feeding 8 encoder and 8 decoder transformer layers) that reports 51 percent native recovery on held-out CATH backbones, almost 10 points over prior methods, rising to 72 percent for buried residues.9 Its central trick was training on roughly 12 million AlphaFold2-predicted structures on top of the experimental CATH set.9 LigandMPNN extends ProteinMPNN to non-protein context, and the gains near functional sites are large: 63.3 percent interface recovery for small molecules (versus 50.5 for ProteinMPNN), 50.5 percent for nucleotides (versus 34.0), and 77.5 percent for metals (versus 40.6).8
Backbone diffusion and flow matching models generate 3D structure, leaving sequence to an inverse folder. RFdiffusion fine-tunes a denoising process onto the RoseTTAFold network and reported beating prior design methods across multiple tasks.4 FrameDiff trains an SE(3) diffusion model from scratch, no pretrained structure network, at about 17.4M parameters; its successor FrameFlow recasts the same frame representation as SE(3) flow matching and reports roughly 2x better designability with about 5x fewer sampling steps.56 Genie 2 scaled training to 588,571 FoldSeek-clustered AFDB structures and reported state-of-the-art designability, diversity, and novelty among compared methods, plus a lead on multi-motif scaffolding.3 Chroma introduced a polymer-aware diffusion process with sub-quadratic scaling and natural-language conditioning, reporting high in-silico designability for samples up to about 1000 residues and complexes up to about 4000.10
SALAD is the interesting recent entry on efficiency. Its sparse attention drops complexity from O(N^2) to roughly O(N*K), and it reports matching RFdiffusion and Genie 2 up to about 400 residues while outperforming them on longer proteins, generating a 1000-residue backbone in about 19 seconds on a single RTX 3090 versus over 10 minutes for RFdiffusion.2 That is roughly 100x faster. At 8 million parameters.2
Sequence-based generation skips structure entirely. EvoDiff runs discrete diffusion over amino-acid sequences (38M and 640M sizes), trained on UniRef50, and its headline contribution is generating intrinsically disordered regions that structure-based methods cannot touch.11 ProGen3 is the scaling-laws play: a sparse Mixture-of-Experts language model trained on Profluent's 3.4-billion-protein, 1.1-trillion-token atlas, deriving compute-optimal scaling curves across 112M to 46B parameters.12
Notice what almost all of these report. FrameFlow, Genie 2, SALAD, and Chroma headline with in-silico metrics. SALAD and the authors of FrameFlow say so explicitly: no wet-lab validation in the core benchmarks.26 These are capability-ledger models. That is not a criticism; backbone generators are research tools meant to feed a pipeline. But it means their headline numbers tell you nothing directly about whether a design will fold on the bench.
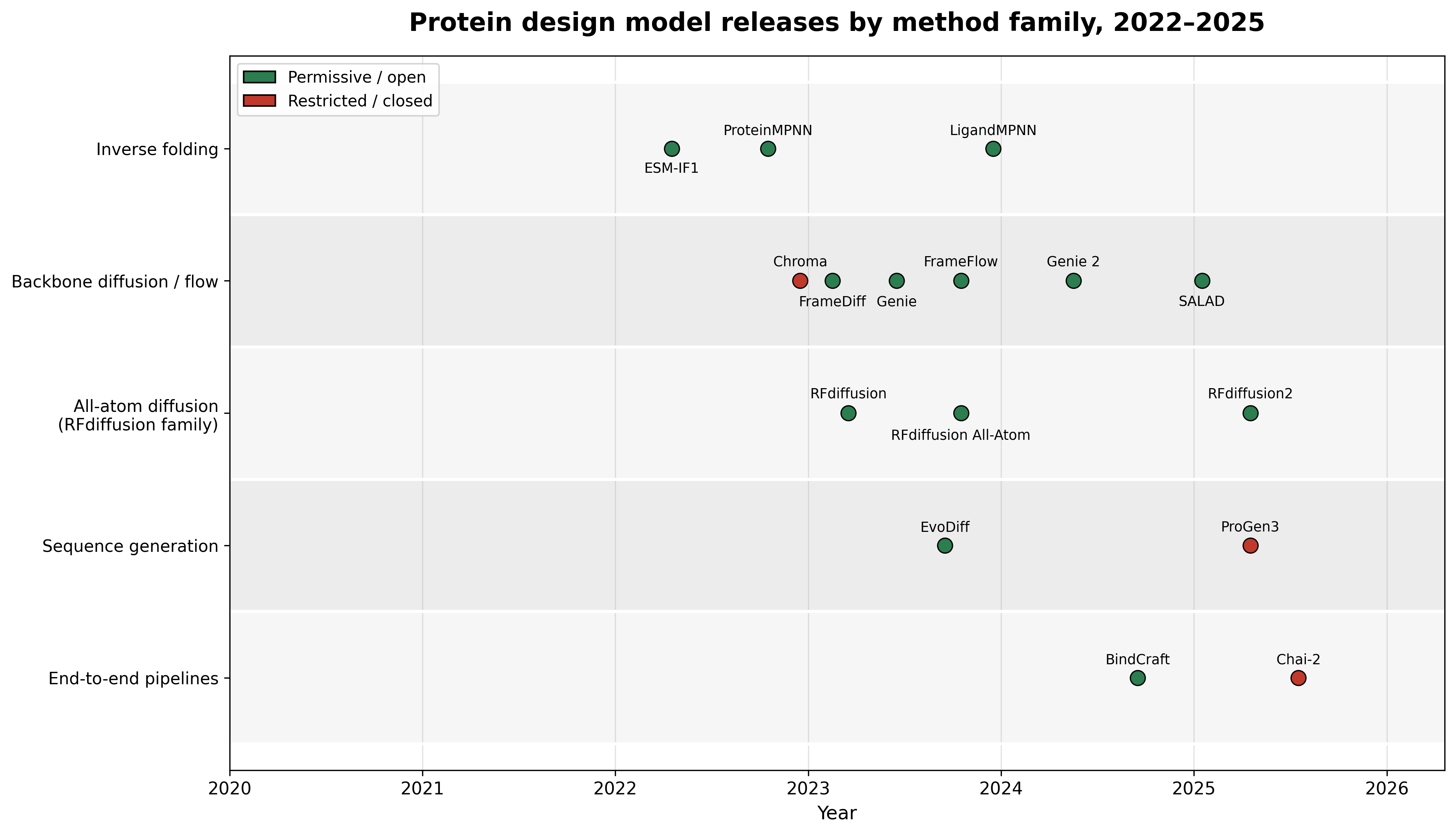 Release timeline by method family. The open foundation was laid in 2022-2023; the 2025 cohort splits, with SALAD and RFdiffusion2 continuing the permissive tradition while ProGen3 (non-commercial) and Chai-2 (closed) arrive with restricted terms. Dates are original release per the source papers: ProteinMPNN,7 ESM-IF1,9 LigandMPNN,8 FrameDiff,5 FrameFlow,6 Genie,16 Genie 2,3 Chroma,10 SALAD,2 RFdiffusion,4 RFdiffusion All-Atom,17 RFdiffusion2,1 EvoDiff,11 ProGen3,12 BindCraft,13 Chai-2.14
Release timeline by method family. The open foundation was laid in 2022-2023; the 2025 cohort splits, with SALAD and RFdiffusion2 continuing the permissive tradition while ProGen3 (non-commercial) and Chai-2 (closed) arrive with restricted terms. Dates are original release per the source papers: ProteinMPNN,7 ESM-IF1,9 LigandMPNN,8 FrameDiff,5 FrameFlow,6 Genie,16 Genie 2,3 Chroma,10 SALAD,2 RFdiffusion,4 RFdiffusion All-Atom,17 RFdiffusion2,1 EvoDiff,11 ProGen3,12 BindCraft,13 Chai-2.14
The validity ledger: what the wet lab says
Now the models that report experimental outcomes, and the numbers that should actually move your decision.
BindCraft is not a trained model at all. It is a pipeline that hallucinates binders by backpropagating gradients through frozen AlphaFold2, then designs sequences with ProteinMPNN and filters with PyRosetta.13 It trains zero new parameters.13 What makes it an anchor for this whole post is the validity number: experimental success rates of 10 to 100 percent across cell-surface receptors, allergens, de novo proteins, and even multi-domain nucleases like CRISPR-Cas9, reaching nanomolar affinities, with hits typically from fewer than 10 designs.1315 It published in Nature in 2025.15 The 10-to-100 percent range is honest and important: success is target-dependent, and the low end of that range is a real outcome you should plan for.
RFdiffusion has the longest experimental track record of the generative backbone models. The Nature 2023 paper reported hundreds of experimentally characterized designs: a picomolar-affinity binder generated by computation alone, symmetric assemblies confirmed by electron microscopy, and metal-binding proteins.4 The lineage kept validating in the lab. RFdiffusion All-Atom designed and experimentally confirmed proteins binding digoxigenin, heme, and bilin.17 RFdiffusion2 is the strongest recent enzyme result: the 41/41-versus-16/41 scaffolding sweep, with three active catalysts found while screening fewer than 96 sequences each, a best design at kcat/KM around 53,000 M^-1 s^-1, and X-ray confirmation.1 That is both ledgers in one paper, which is what good design work looks like.
LigandMPNN reports experimental validation of over 100 small-molecule- and DNA-binding proteins, some with up to roughly 100-fold affinity improvements.8 ProGen3 reports that generated proteins expressed at levels comparable to natural proteins from the same family, and that the 46B model produced 59 percent more diversity than the 3B and 198 percent more than the 339M model at 30 percent sequence identity.12 Chroma reports wet-lab characterization (expression, circular dichroism) for a subset of designs in its paper, though most of its evidence is in-silico.10
Then there is Chai-2, which deserves its own paragraph because it is the model most likely to be over-quoted. The reported numbers are striking: a 16 percent experimental hit rate across 52 novel antibody targets using 20 or fewer candidates per target (20.0 percent for VHHs, 13.7 percent for scFvs), at least one validated binder for 50 percent of targets in a single round, and a claimed greater-than-100x improvement over prior computational methods that typically managed under 0.1 percent.14 A separate, smaller miniprotein evaluation reports about 68 percent hit rate across five targets.14
Here is the discipline the two-ledger habit forces. Those Chai-2 numbers are self-reported in a non-peer-reviewed bioRxiv technical report from the company selling the platform.14 The weights, architecture, parameter count, and training data are all undisclosed.14 No one outside Chai Discovery can reproduce any of it. The 68 percent figure rests on five targets. A "hit" means binding detection, not validated developability or affinity for every hit.14 None of that means the numbers are wrong. It means they are a hypothesis with no path to independent verification, which is a different epistemic object from BindCraft's peer-reviewed Nature result.15 Read them accordingly.
Below is the benchmark table that matters most, deliberately split so the two ledgers never sit in the same column.
| Model | Capability ledger (in-silico) | Validity ledger (wet-lab) |
|---|---|---|
| ProteinMPNN | 52.4% native seq recovery vs 32.9% Rosetta7 | extensive: rescued failed Rosetta/AF designs, X-ray/cryoEM confirmed7 |
| ESM-IF1 | 51% recovery (72% buried residues)9 | none reported by authors9 |
| LigandMPNN | 63.3% / 50.5% / 77.5% interface recovery (small mol / nucleotide / metal)8 | >100 small-molecule & DNA binders, up to ~100x affinity gains8 |
| RFdiffusion | beats prior methods across tasks4 | picomolar binder by computation alone; hundreds characterized4 |
| RFdiffusion All-Atom | competitive structure-prediction accuracy17 | digoxigenin, heme, bilin binders confirmed17 |
| RFdiffusion2 | 41/41 active sites vs 16/41 prior best1 | 3 active catalysts, <96 seqs each, kcat/KM ~53,0001 |
| FrameDiff / FrameFlow | FrameFlow ~2x designability, ~5x fewer steps6 | none reported by authors6 |
| Genie 2 | SOTA designability/diversity/novelty among compared3 | none in core papers3 |
| Chroma | high designability to ~1000 aa, complexes ~4000 aa10 | subset: expression + CD characterization10 |
| SALAD | 36.7% designability at 1000 aa; ~100x faster than RFdiffusion2 | none in core benchmarks (in-silico only)2 |
| EvoDiff | self-consistency foldability; strong IDR generation11 | selected: expressing/folding, metal-binding, IDR signals11 |
| ProGen3 | scaling laws 112M-46B; ProteinGym baseline12 | expression comparable to natural family proteins12 |
| BindCraft | n/a (uses AF2 + ProteinMPNN scoring)13 | 10-100% success, nanomolar, <10 designs1315 |
| Chai-2 | undisclosed14 | 16% antibody hit rate (company-reported, not replicated)14 |
The column on the right is the one that should drive a deployment decision. The column on the left is where you go shopping for a tool to put into a pipeline.
The two-stage workflow, and where each model fits
The field stands on two workhorses, ProteinMPNN and RFdiffusion, and almost everything else is an arrangement around them. The reason is the workflow. For most de novo work the pipeline is two stages plus a filter, and almost no single model does all of it.
Stage one generates a backbone. RFdiffusion, Chroma, FrameDiff, FrameFlow, Genie 2, and SALAD all live here. They output 3D coordinates and stop.410532 FrameDiff is explicit about the consequence: backbone-only, needs a separate inverse-folding model plus structure-prediction validation, with no wet-lab confirmation by the authors.5 That is the norm for this stage, not the exception. The capability story within the stage is mostly about what you are optimizing for. RFdiffusion leverages pretrained RoseTTAFold weights, which is why it tends to lead on raw designability; FrameDiff and FrameFlow train from scratch and sit below it precisely because they do not borrow those weights.456 Genie 2 is the Apache-2.0 academic alternative, strongest when you need to scaffold several motifs at once.3 SALAD is the speed-and-length option.2
| Backbone generator | Designability signal | Standout strength | Validation | License (weights) |
|---|---|---|---|---|
| RFdiffusion4 | outperformed prior methods (2023) | broadest task range; picomolar binder | extensive wet lab4 | BSD4 |
| Genie 23 | SOTA among compared methods | multi-motif scaffolding, AFDB-scale | in-silico3 | Apache-2.03 |
| SALAD2 | 36.7% at 1000 aa; matches peers ≤400 aa | ~100x faster; long proteins | in-silico (ESMFold)2 | CC-BY-4.02 |
| FrameDiff / FrameFlow56 | FrameFlow ~2x over FrameDiff, ~5x fewer steps | lightweight, no pretrained net | in-silico56 | MIT5 |
| Chroma10 | high designability to ~1000 aa | programmable + text-prompted | mostly in-silico10 | non-commercial weights10 |
Stage two designs a sequence for that backbone. This is inverse folding, and it is the most mature, most validated, most permissively licensed part of the field. ProteinMPNN, LigandMPNN, and ESM-IF1 do this.789 If your backbone has a bound ligand, nucleic acid, or metal, you want LigandMPNN, because the original ProteinMPNN is not aware of non-protein atoms and underperforms near functional sites, which was the entire motivation for the successor.8
| Inverse-folding model | Sequence recovery | Best for | License |
|---|---|---|---|
| ProteinMPNN7 | 52.4% overall (vs 32.9% Rosetta) | protein-only backbones | MIT7 |
| LigandMPNN8 | 63.3% small-mol / 50.5% nucleotide / 77.5% metal (interface) | anything with ligands, nucleic acids, metals | MIT8 |
| ESM-IF19 | 51% overall (72% buried) on CATH | general single-chain, AF2-augmented, log-likelihood scoring | MIT (repo archived)9 |
Then you filter in silico: fold the designed sequence with AlphaFold2 or ESMFold, check self-consistency, throw away the failures, and only then order DNA. That filtering step is the capability ledger doing its job as triage, not as a verdict. Nothing goes to the wet lab unscreened. This is the field's version of "always beat a trivial baseline first."
A few models collapse the stages. Chroma co-generates backbone and sequence in one correlated diffusion process plus a design network.10 EvoDiff and ProGen3 are sequence-only: they skip backbone generation, which is exactly why EvoDiff can produce disordered regions that have no fixed backbone to design against, but they then need a downstream folder to assess structure.1112 BindCraft and Chai-2 are the end-to-end pipelines, generating and scoring binders against a target in one loop, which is what makes their wet-lab numbers comparable to each other and incomparable to a bare backbone generator.1314
The licensing ledger: open, restricted, closed
For an engineer, license is not a footnote. It decides whether you can ship, and it is the first filter, not the last. Here is the table to read if you are choosing a model to deploy rather than to cite. For each model it states the license that governs the artifact you would actually run, and whether you can use it commercially without a separate agreement.
| Model | Code license | Weights / artifact license | Commercial deploy without extra license? | Notes |
|---|---|---|---|---|
| ProteinMPNN | MIT7 | MIT (weights included)7 | Yes | both code and weights MIT, no NC clause7 |
| LigandMPNN | MIT8 | MIT (weights included)8 | Yes | code and parameters both MIT8 |
| ESM-IF1 | MIT9 | MIT (weights included)9 | Yes | entire facebookresearch/esm repo is MIT9 |
| EvoDiff | MIT11 | MIT11 | Yes | permissive, attribution only11 |
| FrameDiff / FrameFlow | MIT5 | MIT5 | Yes | READMEs say research-only; MIT terms are not NC5 |
| Genie / Genie 2 | Apache-2.03 | Apache-2.03 | Yes | no NC restriction observed3 |
| SALAD | Apache-2.02 | CC-BY-4.02 | Yes | attribution only; commercial allowed2 |
| RFdiffusion | BSD4 | BSD (covers weights)4 | Yes | UW BSD, for-profit and non-profit allowed4 |
| RFdiffusion All-Atom | BSD17 | BSD17 | Yes | no NC restriction17 |
| RFdiffusion2 | BSD-3-Clause1 | BSD-3-Clause1 | Yes | some write-ups wrongly say MIT; LICENSE is BSD-31 |
| BindCraft | MIT15 | n/a (no new weights)15 | No, gated by PyRosetta | MIT code, but default pipeline needs a paid PyRosetta commercial license15 |
| ProGen3 | Apache-2.012 | CC-BY-NC-SA-4.012 | No | released weights non-commercial; ≤3B open, 46B unreleased12 |
| Chroma | Apache-2.010 | custom non-commercial10 | No | weights research-only, gated by API key; commercial needs Generate Biomedicines license10 |
| Chai-2 | none (closed)14 | not released14 | No | proprietary; access via paid partnership only14 |
Ten of the 14 are clean for commercial deployment, and notably that includes the entire Baker Lab output. That is the good news, and it is why protein design feels open. The four that are not split into three distinct failure modes, and the distinction is worth internalizing because each one bites differently.
The dependency trap. BindCraft's repository carries an MIT license, which is true and also misleading.15 BindCraft is not a trained model; it is an automated pipeline that hallucinates binders by backpropagating through frozen AlphaFold2, then uses ProteinMPNN for sequence design and PyRosetta for relaxation, scoring, and filtering.15 PyRosetta is free for academic and non-commercial use but requires a separate paid commercial license from the University of Washington.15 So the default pipeline's commercial path runs straight into a license fee that the MIT badge never mentions. A community fork, FreeBindCraft, exists specifically to remove the mandatory PyRosetta dependency.15 If you cloned BindCraft expecting MIT to mean free, check your dependency tree before you ship.
The non-commercial weights trap. ProGen3 releases its code under Apache 2.0 but its weights under CC BY-NC-SA 4.0, non-commercial and ShareAlike, with a second catch layered on top: only the sizes up to 3B have openly downloadable weights, while the 6B and 46B flagships described in the paper are not released.12 Chroma is the same shape on the weights side. Its code is Apache 2.0, but the parameters carry a custom non-commercial "Chroma Parameters License" granted only to academics and non-profits, with weight download gated behind API key and registration; commercial use requires a separate license from Generate Biomedicines.10
The fully closed model. Chai-2 has no public or open-source license, no downloadable weights, no public API, and no public web server.14 Access is by application to Chai Discovery with paid annual fees and partnership agreements, and the model is embedded in commercial deals with Pfizer and Eli Lilly, sometimes trained on partners' proprietary data.14 Notably, the predecessor Chai-1 was released openly, so Chai-2 represents a deliberate move toward closure within a single company's lineage.14
If commercial freedom and reproducibility are hard constraints, the Baker Lab stack plus SALAD, Genie 2, ESM-IF1, and EvoDiff covers nearly the whole design pipeline with permissive licenses and published, peer-reviewed validation.
Choosing by task: the decision tree
Decisions, not leaderboards. The fastest way to act on all of the above is to ask what you are designing, then let the answer narrow you to one or two models. Five branches cover the field.
 A task-first router for the 14 models. Each leaf narrows to one or two choices; license color-coding flags what you can ship. Sources: ProteinMPNN,7 LigandMPNN,8 ESM-IF1,9 RFdiffusion,4 Genie 2,3 SALAD,2 FrameFlow,6 BindCraft,1315 Chai-2,14 RFdiffusion All-Atom,17 RFdiffusion2,1 Chroma,10 EvoDiff,11 ProGen3.12
A task-first router for the 14 models. Each leaf narrows to one or two choices; license color-coding flags what you can ship. Sources: ProteinMPNN,7 LigandMPNN,8 ESM-IF1,9 RFdiffusion,4 Genie 2,3 SALAD,2 FrameFlow,6 BindCraft,1315 Chai-2,14 RFdiffusion All-Atom,17 RFdiffusion2,1 Chroma,10 EvoDiff,11 ProGen3.12
You have a backbone and need a sequence. Use ProteinMPNN. It is MIT, about 1.7M parameters, runs anywhere, and is the most validated option.7 If your design sits near a small molecule, nucleotide, or metal, switch to LigandMPNN, which is also MIT and recovers interface residues far better near non-protein atoms.8 ESM-IF1 is a reasonable third option, also MIT, when you want a transformer-based alternative or its log-likelihood scoring; note its parent repository is archived read-only, so expect no maintenance.9
You need a de novo backbone. RFdiffusion is the default, BSD-licensed, with the strongest experimental track record.4 If you need long proteins fast, SALAD generates 1000-residue backbones roughly 100x faster than RFdiffusion and is commercial-friendly, with the caveat that designability drops to 36.7 percent at 1000 residues and the implementation is JAX-only.2 Genie 2 is the strong Apache-2.0 academic alternative, especially for multi-motif scaffolding.3 FrameDiff and FrameFlow are for understanding the SE(3) generative recipe, not for shipping designs.56
You need a binder against a target. In academia, BindCraft delivers one-shot functional binders, often from fewer than 10 designs, with reported wet-lab success of 10 to 100 percent.13 At a company, audit the PyRosetta dependency first or use FreeBindCraft.15 The open primitive underneath is RFdiffusion plus ProteinMPNN, the same picomolar-binder pipeline.47 If you need antibody design and have a budget and a partnership, Chai-2 is the only one of these built specifically for that, with the caveats above.14
You are designing around a small molecule, nucleic acid, or metal. RFdiffusion All-Atom for small-molecule binders, validated on digoxigenin, heme, and bilin.17 RFdiffusion2 for enzymes designed from a catalytic mechanism, the only model here that scaffolds atom-level active sites directly.1 LigandMPNN for the sequence step on any of these all-atom backbones.8 Chroma if you want programmable conditioning and can live with non-commercial weights.10
You need de novo sequences without a target structure. EvoDiff for disordered regions or a fully MIT-licensed, commercially deployable option.11 ProGen3 for large-scale diverse generation, but only academically given the non-commercial weights and the 3B cap on open checkpoints.12
What is not known
Being explicit about gaps is part of the job, and it belongs in the open, not quarantined.
Parameter counts are undisclosed for a surprising number of these: Chroma, Genie, the entire RFdiffusion family, LigandMPNN, and Chai-2 all report "unknown" or decline to state.10164171814 You can infer rough scale (the RFdiffusion family sits on RoseTTAFold-class networks of tens of millions of parameters, and the later RFdiffusion3 reports 168M), but the exact figures are not public.1718
Cross-model wet-lab comparisons do not exist. Every experimental number in the validity ledger was generated by a different lab, on different targets, with different success criteria. BindCraft's 10-to-100 percent, Chai-2's 16 percent, and RFdiffusion2's three validated catalytic sites are not measuring the same thing on the same panel. There is no shared experimental benchmark, so the validity ledger is a set of anecdotes from credible sources, not a leaderboard.
Most backbone generators have no author-reported wet-lab validation at all. FrameDiff, FrameFlow, Genie 2, and SALAD report in-silico designability only.5632 Their real-world success rate is genuinely unknown, and you should treat their headline numbers as the capability hypotheses they are.
Chai-2's entire profile is undisclosed and unreplicated.14 Until an independent group publishes a replication, every claim about it is provisional.
The bottom line
Read this literature with two ledgers open. The capability ledger, full of designability and self-consistency scores, tells you which tool to put in a pipeline. It is cheap, abundant, and only loosely connected to reality. The validity ledger, full of binder hit rates and crystal structures, tells you what actually works, and it is sparse, expensive, and scattered across incompatible experiments. When the two disagree, trust the wet lab. When only the capability ledger has an entry, treat the number as a hypothesis. And when a striking validity number comes from a closed model that no one can reproduce, file it under "promising, unverified," not "state of the art."
The good news for practitioners is that the most validated tools are also the most open. You can build a complete, commercially deployable stack from MIT and BSD pieces: RFdiffusion or SALAD for backbones, ProteinMPNN or LigandMPNN for sequences, ESM-IF1 in reserve, AlphaFold2 or ESMFold for the filter, all free to ship.42789 The closed and non-commercial models (Chai-2, Chroma, and ProGen3's weights) are not better enough to justify their terms for most teams, and the one that looks free, BindCraft, is not, unless you handle the PyRosetta dependency.14101215 Start there, beat a trivial baseline first, and put your own designs in a real plate before you believe any benchmark, including your own. In this field, more than most, read the license before the benchmark. The license is the spec.
References
Footnotes
-
Ahern, Yim, Tischer, ... Baker et al., "Atom-level enzyme active site scaffolding using RFdiffusion2," Nature Methods 23:96-105 (2026). https://doi.org/10.1038/s41592-025-02975-x ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16
-
Jendrusch, M. & Korbel, J.O. "Efficient protein structure generation with sparse denoising models," Nature Machine Intelligence 7, 1429-1445 (2025). https://doi.org/10.1038/s42256-025-01100-z ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20 ↩21 ↩22 ↩23 ↩24 ↩25 ↩26 ↩27
-
Lin, Lee, Zhang & AlQuraishi, "Out of Many, One: Designing and Scaffolding Proteins at the Scale of the Structural Universe with Genie 2," arXiv:2405.15489 (2024). https://arxiv.org/abs/2405.15489 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19
-
Watson, J. L. et al., "De novo design of protein structure and function with RFdiffusion," Nature 620, 1089-1100 (2023). https://www.nature.com/articles/s41586-023-06415-8 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20 ↩21 ↩22 ↩23 ↩24
-
Yim et al., "SE(3) diffusion model with application to protein backbone generation," ICML 2023, arXiv:2302.02277. https://arxiv.org/abs/2302.02277 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20
-
Yim et al., "Fast protein backbone generation with SE(3) flow matching," arXiv:2310.05297. https://arxiv.org/abs/2310.05297 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13
-
Dauparas et al., "Robust deep learning-based protein sequence design using ProteinMPNN," Science 378:49-56 (2022). https://www.science.org/doi/10.1126/science.add2187 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20 ↩21
-
Dauparas J. et al., "Atomic context-conditioned protein sequence design using LigandMPNN," Nature Methods 22, 717-723 (2025). https://www.nature.com/articles/s41592-025-02626-1 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20 ↩21 ↩22 ↩23
-
Hsu et al., "Learning inverse folding from millions of predicted structures," ICML 2022. https://proceedings.mlr.press/v162/hsu22a.html ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20
-
Ingraham, Baranov, Costello et al., "Illuminating protein space with a programmable generative model," Nature 623:1070-1078 (2023). https://doi.org/10.1038/s41586-023-06728-8 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20 ↩21 ↩22 ↩23 ↩24
-
Alamdari et al., "Protein generation with evolutionary diffusion: sequence is all you need," bioRxiv 2023. https://www.biorxiv.org/content/10.1101/2023.09.11.556673 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16
-
"Scaling unlocks broader generation and deeper functional understanding of proteins" (Profluent), bioRxiv 2025. https://www.biorxiv.org/content/10.1101/2025.04.15.649055v2.full ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20
-
BindCraft preprint: "One-shot design of functional protein binders with BindCraft," bioRxiv 2024. https://www.biorxiv.org/content/10.1101/2024.09.30.615802 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13
-
"Zero-shot antibody design in a 24-well plate," Chai Discovery Team, bioRxiv 2025. https://www.biorxiv.org/content/10.1101/2025.07.05.663018v1 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20 ↩21 ↩22 ↩23 ↩24 ↩25 ↩26
-
Pacesa, M. et al. "One-shot design of functional protein binders with BindCraft," Nature (2025). https://doi.org/10.1038/s41586-025-09429-6 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15
-
Lin & AlQuraishi, "Generating Novel, Designable, and Diverse Protein Structures by Equivariantly Diffusing Oriented Residue Clouds," ICML 2023, arXiv:2301.12485. https://arxiv.org/abs/2301.12485 ↩ ↩2 ↩3 ↩4 ↩5
-
Krishna, Wang, Ahern et al., "Generalized biomolecular modeling and design with RoseTTAFold All-Atom," Science (2024). https://www.science.org/doi/10.1126/science.adl2528 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15
-
"De novo design of all-atom biomolecular interactions with RFdiffusion3," bioRxiv 2025. https://www.biorxiv.org/content/10.1101/2025.09.18.676967 ↩ ↩2
Comments and feedback
Spotted an error or have a counterpoint? Comment below. No account needed, a name is enough. Corrections and pushback are welcome.