In 2024, a model called ESM3 produced a working green fluorescent protein whose sequence was only about 58% identical to the nearest known fluorescent protein, a distance the authors estimated as equivalent to roughly 500 million years of natural evolution.1 The protein folded. It fluoresced. The authors named it esmGFP.
That number, 58%, is the kind of result that ends up in keynote slides. It is also a good place to start being careful, because it tells you something real about generative capability and almost nothing about whether you should swap your current pipeline for the model that produced it.
It helps to keep two ledgers while reading this literature. One ledger records capability: what a model can demonstrably do under some conditions, often the conditions its authors chose. The other records validity: what survives a held-out test set, an honest baseline, and a license you can actually deploy under. A protein language model can score brilliantly on the first ledger and still be the wrong tool on the second. Most of the confusion in this field comes from collapsing the two.
This is a survey of the 18 models that the rewire.it Foundation Models in Biology catalog classifies as protein language models. I will keep both ledgers open throughout: a quick decision tree for the impatient, an architecture taxonomy, the two ledgers in detail, the licensing fault line that decides what you can ship, and recommendations by task.
What a protein language model actually is
A protein language model (PLM) borrows the self-supervised recipe from natural-language processing and applies it to amino-acid sequences. The dominant pattern is a transformer encoder trained with masked language modeling, BERT-style: hide some residues, predict them from context. This is the lineage that runs through ESM-1b,2 ESM-2,3 and ESM C.4 The other major pattern is autoregressive, decoder-only, next-token prediction, which is what ProGen and ProGen2 use56 and what makes them natural generators.
The output you usually care about is not the predicted token. It is the internal representation, the embedding, which you feed into a downstream head for some task you have labels for. The finding that launched the field was that these representations are not arbitrary. Rives and colleagues reported that secondary structure and long-range residue contacts emerge in the attention maps of a model trained only to fill in masked residues, with features competitive with HMM profiles for supervised secondary-structure prediction.2
The fast answer: a decision tree by task
Most readers want the decision before the reasoning. Here it is as a tree. The sections that follow defend each branch.

The single most important branch is not on the diagram. It is the license fork that cuts across every task: ESM-2 is MIT and commercially usable,3 while ESM3 and ESM C 600M are non-commercial research-only.14 If you are in industry or building a clinical product, that fork eliminates a large fraction of the "best" models before you benchmark anything.
The taxonomy: four architectures plus a CNN holdout
Almost every protein language model descends from one of a few text-model architectures, repurposed to treat amino acids, and sometimes structure tokens, as a vocabulary. The architecture tells you, more than anything else, what the model is for.

That diagram is most of the taxonomy in one view: how a model tokenizes its input and which direction it predicts. Almost every practical difference between these 18 models falls out of those two choices.
Encoder, masked-LM models are your representation workhorses. ESM-1b set the template, a 33-layer encoder with embedding dimension 1280 and about 650M parameters, trained on UniRef50.2 ESM-2 kept the recipe, swapped learned positional embeddings for rotary embeddings, and scaled across six sizes from 8M to 15B.3 ProtBERT mirrors BERT-Large dimensions at about 420M.7 AMPLIFY modernized the recipe with RMSNorm, SwiGLU, and rotary embeddings, in two open sizes of 120M and 350M.8 ESM C, the EvolutionaryScale successor for embeddings, uses pre-LayerNorm, RoPE, and SwiGLU at 300M, 600M, and 6B.4
Decoder, autoregressive models are your generators. ProGen pioneered control-tag conditioning, CTRL-style, as a roughly 1.2B-parameter conditional transformer.5 ProGen2 scaled the idea to a 151M-through-6.4B suite, trained both left-to-right and right-to-left for N-to-C and C-to-N generation.6 ProtGPT2 is a clean GPT2-large adaptation at 738M, with a BPE tokenizer where each token averages about four amino acids.9 ProLLaMA took the LLM-native route: continual pretraining of LLaMA-2-7B with LoRA and instruction tuning, which is also why its license is a headache.10
Encoder-decoder, structure-aware, and multimodal models are where the field is moving. ProtT5 is a T5 encoder-decoder at roughly 3B (XL) and 11B (XXL), of which in practice only the encoder is used for embeddings.7 Ankh is a deliberately small T5-family model, optimized through more than 20 ablation experiments, with a base around 450M.11 ProstT5 and SaProt both inject structure: ProstT5 fine-tunes ProtT5-XL-U50 to translate between amino acids and Foldseek 3Di tokens,12 while SaProt fuses each residue token with a 3Di structure token on the ESM-2 architecture.13 ESM3 and xTrimoPGLM are the unification attempts: ESM3 fuses sequence, structure, and function as discrete tracks,1 and xTrimoPGLM uses a General Language Model backbone to unify masked understanding and autoregressive generation in one 100B model.14 PoET sits slightly apart as the retrieval-augmented family model.15
The CNN holdout. CARP is the contrarian: a dilated 1D convolutional encoder based on ByteNet, not a transformer, that scales linearly with sequence length and uses no positional embeddings, so it generalizes to sequences longer than those seen in training.16 Trained on the same March 2020 UniRef50 as ESM-1b with the same masked-LM objective, CARP-640M is competitive with, and occasionally superior to, ESM-1b across downstream tasks.16 The CARP result matters as an existence proof that the transformer is not load-bearing for protein masked language modeling; the inductive bias of masked prediction on UniRef is.
The master comparison
Here is the full field. Read it as a scan, not a ranking. The license column is the one most people get wrong, and it is the one that decides whether you can ship.
| Model | Family | Released | Params (largest open) | License (weights) | Commercial? |
|---|---|---|---|---|---|
| ESM-1b | Encoder/MLM | 20192 | ~650M2 | MIT2 | Yes2 |
| ESM-1v | Encoder/MLM | 202117 | 650M x5 ensemble17 | MIT17 | Yes17 |
| ESM-2 | Encoder/MLM | 20223 | 15B (650M most used)3 | MIT3 | Yes3 |
| ESM3 | Multimodal | 2024.061 | 1.4B open (98B API)1 | Cambrian Non-Commercial1 | No1 |
| ESM C | Encoder/MLM | 2024.124 | 300M open (6B API)4 | Tiered Cambrian4 | Mixed4 |
| ProGen | Decoder/AR | 20205 | ~1.2B5 | BSD-35 | Yes5 |
| ProGen2 | Decoder/AR | 2022.066 | 6.4B6 | BSD-36 | Yes6 |
| ProtGPT2 | Decoder/AR | 20229 | 738M9 | Apache-2.09 | Yes9 |
| ProLLaMA | Decoder/AR | 2024.0210 | ~7B (LLaMA-2)10 | Llama-2 Community10 | Restricted10 |
| ProtBERT | Encoder/MLM | 20207 | ~420M7 | AFL-3.07 | Yes7 |
| ProtT5 | Enc-Dec/T5 | 20207 | ~11B (XXL)7 | AFL-3.0 weights7 | Yes7 |
| Ankh | Enc-Dec/T5 | 2023.0111 | ~2B (Ankh2)11 | CC-BY-NC-SA-4.011 | No11 |
| ProstT5 | Enc-Dec/3Di | 2023.0712 | ~3B12 | MIT12 | Yes12 |
| SaProt | Encoder/3Di-fused | 2023.1013 | 1.3B13 | MIT13 | Yes13 |
| CARP | CNN/MLM | 202216 | ~640M16 | BSD-style16 | Yes16 |
| AMPLIFY | Encoder/MLM | 2024.098 | 350M8 | MIT8 | Yes8 |
| PoET / PoET-2 | Seq-of-seqs | 2023 / 20251518 | 182M (PoET-2)18 | Non-Commercial15 | No15 |
| xTrimoPGLM | Unified GLM | 2023.0714 | 100B (INT4 open)14 | CC-BY-NC-4.014 | No14 |
A few patterns are worth naming before we go deeper. The most-cited, most-deployed models (the ESM line through ESM-2, plus ProGen2, ProtGPT2, SaProt, ProstT5, AMPLIFY) are permissively licensed. The frontier-scale and structure-aware-generative models (ESM3, ESM C 600M and 6B, Ankh, xTrimoPGLM, PoET) are frequently non-commercial. That correlation is not an accident, and it shapes what most labs can actually use.
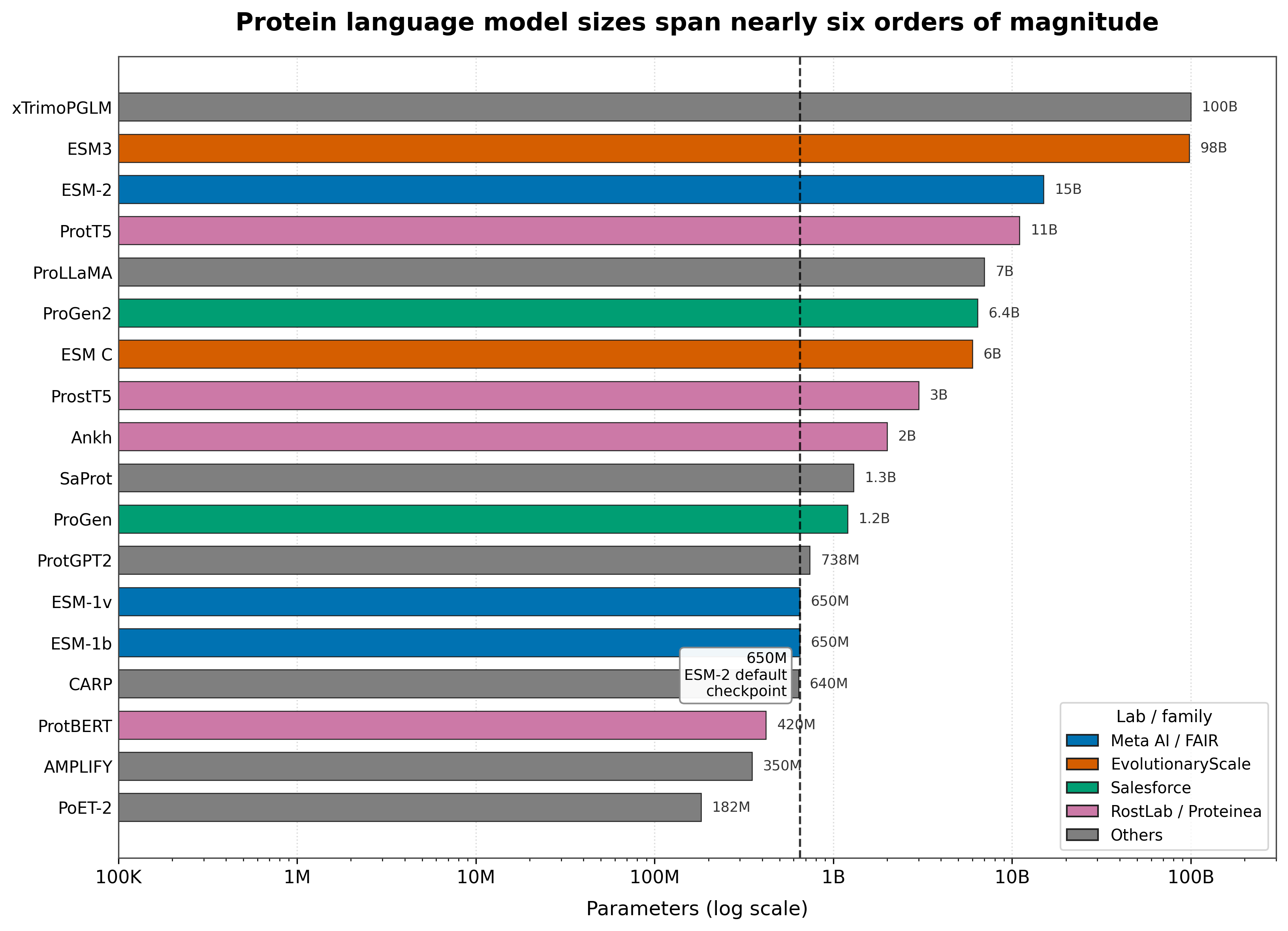
Figure: largest openly released parameter count per model, log scale. Sizes span nearly six orders of magnitude, from PoET-2 at 182M18 to xTrimoPGLM's INT4 100B checkpoint.14 The most-downloaded checkpoint in practice is still ESM-2 650M.3
Capability ledger: the three headline claims
Three claims do most of the marketing work in this field. Each is true in a specific sense. Each needs an asterisk.
Claim 1: zero-shot variant effect prediction
The cleanest capability result in the whole field belongs to ESM-1v. On a benchmark of about 41 deep mutational scanning datasets, ESM-1v achieved zero-shot variant-effect prediction comparable to state-of-the-art supervised and MSA-based methods such as DeepSequence and EVmutation, with no task-specific training and no MSA.17 You take a wild-type sequence, mask each position, read off the model's log-probabilities for the mutant versus wild-type residue, and you have a fitness proxy. No labels. That is genuinely useful, and it is why ESM-1v remains a standard baseline years on.17
The asterisk is the regime. ESM-1v ships as a five-model ensemble, so the best results cost five forward passes, and it is single-sequence.17 Zero-shot variant scoring works well where evolutionary signal is dense and degrades where it is sparse, which is exactly the gap retrieval-augmented models target on the validity ledger below.
Claim 2: structure emerges inside the model
ESM-2 made the emergence story concrete. Performance scales smoothly with parameters from 8M to 15B, reducing masked-LM perplexity and improving unsupervised contact prediction along the way.3 Then ESMFold, the structure head built on ESM-2 15B, reached a TM-score of about 0.71 on CAMEO and was competitive with AlphaFold2 and RoseTTAFold on many targets while running roughly 6x to 60x faster, because it needs no MSA.3
The asterisk lives in that "6x to 60x faster" range and in what the speed costs you. Speed without an MSA is the trade: you gain throughput, you lose accuracy precisely where structure is hardest to predict. For a metagenomic atlas of hundreds of millions of proteins, that trade is obviously worth it. For a single high-stakes target where you already have an alignment, it may not be.
Claim 3: generation of novel functional proteins
This is esmGFP. ESM3 was trained as a bidirectional masked generative transformer with a single unified latent space, where sequence, structure, and function are each tokenized into discrete alphabets and fused as tracks, with structure encoded via a learned VQ-VAE-style tokenizer with geometric attention.1 It was trained on roughly 2.78 billion sequences, 236 million structures, and 539 million function annotations, with the largest model consuming about 771 billion tokens at roughly 1.07e24 FLOPs.1 Out of that came esmGFP, at 58% identity to the nearest natural fluorescent protein.1
ProGen made a quieter but equally important version of this claim earlier. The original ProGen generated artificial lysozymes with catalytic efficiencies comparable to natural enzymes, with 73% of generated proteins functional versus 59% of natural ones, and one AI-designed protein's crystal structure experimentally solved.5 That is a wet-lab validation, not a benchmark.
The asterisk on generation is the largest of all: generative outputs require experimental validation, and esmGFP is a success story selected from a generative process.1 The denominator, how many designs were synthesized to get there, is the number that actually predicts your lab's hit rate, and it is rarely the headline.
Validity ledger: where the headlines bend
Now the other ledger. Four recurring issues separate capability from deployable validity.
MSA-depth dependence. Single-sequence models look MSA-free, but their accuracy still tracks evolutionary information density. PoET was built around this. It models whole protein families as a "sequence-of-sequences," a retrieval-augmented autoregressive transformer that attends order-invariantly across family members.15 PoET-2, at just 182M parameters, reportedly beats the ESM series and ProGen2 on ProteinGym by a large margin, with roughly a 50% improvement on challenging low-homolog proteins and significantly better perplexity than ESM-2 and ProGen2 on proteins with fewer than 10 homologs, despite being up to 80x smaller.18 Read that carefully: the gains concentrate exactly where single-sequence models are weakest. That is MSA-depth dependence showing up as a benchmark gap.
Perplexity is not utility. The cleanest demonstration is AMPLIFY. Its authors titled the paper "Protein Language Models: Is Scaling Necessary?"8 and reported that AMPLIFY 350M matches or surpasses ESM2-15B on several sequence-only tasks despite roughly 43x fewer parameters and 17x fewer training FLOPs.8 ProGen2's authors found the same thing from the other direction: larger models do not consistently outperform smaller ones on fitness prediction.6 The Ankh paper makes the point about data rather than scale, reporting that curated, deduplicated UniRef50 outperformed the larger UniRef100 and BFD corpora because of higher quality and lower redundancy.11 Lower masked-LM perplexity is a smooth function of parameters,3 but downstream task performance is not. Scale helps contact and structure prediction smoothly; on fitness and variant effect it inverts. A benchmark number is a hypothesis about real-world performance, not a measurement of it.
Benchmark validity versus clinical validity. Most reported numbers in this field are self-reported by the model authors on benchmarks the authors chose. ESM C's headline efficiency claims come from the EvolutionaryScale release blog, not a peer-reviewed paper.4 AMPLIFY's comparative numbers come from the developers' own benchmarks.8 xTrimoPGLM reports state-of-the-art on 18 understanding benchmarks across four categories, again largely self-reported.14 None of this is dishonest. It is just not the same as an independent held-out evaluation, and it is very far from clinical validity. PoET-2's inclusion of clinical variant effect prediction in its task list is notable precisely because most PLM evaluations stop at ProteinGym.18
Cost per useful prediction. This is the metric the leaderboards never show. xTrimoPGLM's 100B model is released publicly only as an INT4-quantized checkpoint, and ESM3's 7B and 98B models are API-only.141 Against that, ESM-2 650M runs on a single modest GPU, is MIT-licensed, and is the most widely used checkpoint in the family for a reason.3 For most embedding and variant-scoring pipelines, the well-tuned 650M model wins on cost per useful prediction.
The shape of the field, 2019 to 2025
Those four failure modes did not arrive all at once. They tracked the field's center of gravity as it moved from one lab to many.
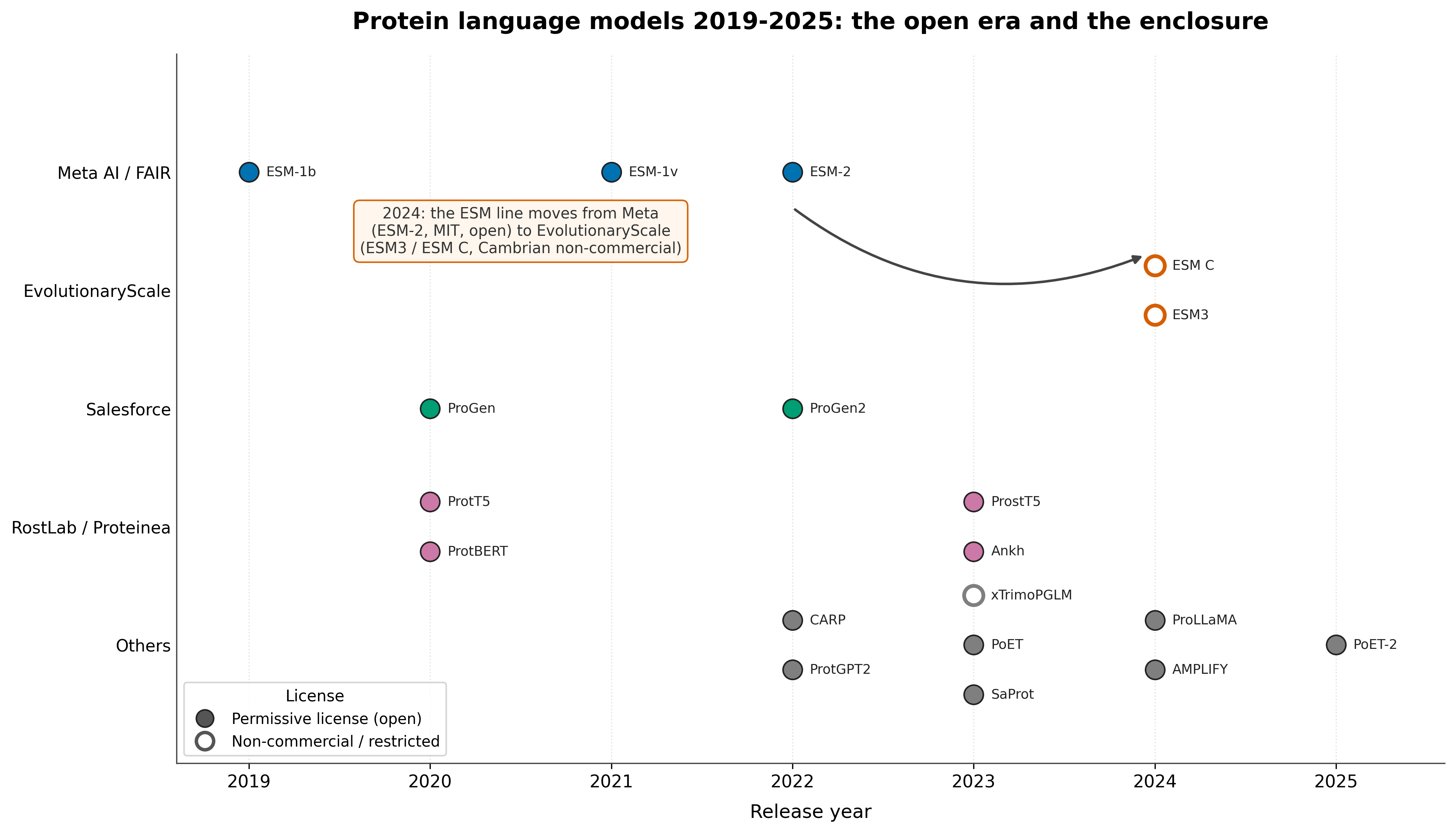
Figure: PLM release timeline, 2019 to 2025, grouped by developer with filled markers for permissive licenses and hollow markers for restricted ones. The field's center of gravity moved from Meta AI / FAIR (2019 to 2022) toward EvolutionaryScale, academic labs, and Chinese and Middle Eastern institutions (2023 onward), and the restricted-license markers cluster after that shift. Release dates and license terms per the model papers and repositories.4231
Choosing by task: the five branches
The decision tree at the top set out five branches. Here is the defense of each, with a "fast answer" before the reasoning. Architecture predicts fit, so choose by what you are actually trying to do.
Branch 1: embeddings for a downstream predictor
You have a labeled dataset, you want frozen features, you will train a small head on top. The right question is which embedding gives your head the most signal per dollar.
The default remains ESM-2 650M: MIT-licensed, widely benchmarked, runs on a single GPU, and the checkpoint most downstream tooling expects.3 One caveat for planners: the official facebookresearch/esm repository was archived read-only on August 1, 2024.3 The weights still work; the lab moved on. The interesting challenger is AMPLIFY 350M, MIT and modern, which reports parity with ESM2-15B at a fraction of the cost, a cheap bet to test on your own task.8 ESM C 300M is the newest efficiency play, but only the 300M tier is open and commercial; the 600M weights are research-only and the 6B is paid API.4 For very long sequences, CARP-640M is a reasonable BSD-style baseline that scales linearly.16
| Use case | Model | Size | License | Why |
|---|---|---|---|---|
| Default open embedding | ESM-2 | 650M3 | MIT3 | Widely benchmarked, single GPU, commercial |
| Cheaper drop-in | AMPLIFY | 350M8 | MIT8 | Reports parity with ESM2-15B, high throughput |
| Newer efficiency | ESM C | 300M4 | Cambrian Open4 | Reported parity with ESM2-650M; 600M is NC |
| Very long sequences | CARP | 640M16 | BSD-style16 | Linear scaling, no positional embeddings |
| Structure-rich data | SaProt | 650M13 | MIT13 | 3Di structure tokens, see Branch 4 |
Branch 2: zero-shot variant effect prediction
You have no labels and want to score mutations directly from a wild-type sequence. ESM-1v is the canonical baseline, MIT, validated on roughly 41 deep mutational scanning datasets.17 ESM-2 can be used the same way and is the more common modern backbone.3 For the hard cases, low-homolog proteins and clinical variants, PoET-2 reports the largest gains, but its weights are non-commercial, so it is a research-only choice unless you license it.1815 SaProt is the permissive structure-aware alternative when you have structures.13 One rule applies across all of them: always beat a trivial baseline first. Position-specific conservation from a simple alignment is often a stubborn competitor.
Branch 3: de novo sequence generation
You want a model to write new sequences, not score existing ones. ProGen2 is the open, commercially usable default, BSD-3, with sizes up to 6.4B.6 Its predecessor ProGen carries the strongest wet-lab validation in the branch, with functional artificial lysozymes.5 ProtGPT2 is the small, Apache-2.0, teaching-and-prototyping option; in its paper, 87.59% of generated sequences were predicted globular versus 88.40% for natural ones.9 The one to flag is ProLLaMA: it reports superfamily prediction exact-match rates of 62% to 67.1%, but its Hugging Face card carries an apache-2.0 tag while the same card and repo state the model follows the Llama 2 license, so the binding terms are the Meta Llama 2 Community License, which is not standard open source.10 If you saw "apache-2.0" and stopped reading, you would be wrong about what you are allowed to do.
Branch 4: structure-aware representation
Sequence-only models leave structure on the table. SaProt is the cleanest design: an ESM-2-style encoder whose vocabulary fuses each residue with a Foldseek 3Di token, reported to surpass ESM-2, ESM-1v, and MIF-ST across 10 downstream tasks, and MIT-licensed for both code and weights.13 ProstT5 takes a translation angle, predicting 3Di from sequence about three orders of magnitude faster than full structure prediction, roughly 43.5 seconds on GPU versus about 48 hours of ColabFold for a benchmark set, with predicted 3Di reaching remote-homology ROC-AUC near 0.45, close to experimental structures at 0.49 and far above pure sequence search at 0.06.12 It is MIT.12 ESM3 handles structure as one of three unified tracks, but it gets its own discussion below because its license defines the central fault line of this survey.1
Branch 5: the largest models
If you want maximum capability regardless of size, you arrive at two giants and a hard licensing wall. ESM3 spans 1.4B (open), 7B, and 98B (API only), and its esmGFP result is the headline of the field.1 xTrimoPGLM reaches 100B parameters, released publicly only as an INT4-quantized checkpoint, and reports state-of-the-art on most of 18 understanding benchmarks, in Nature Methods.14 Both are non-commercial.114 The pattern is hard to miss: the largest, newest, most capable PLMs are disproportionately non-commercial, which is the subject of the next section.
Benchmark table: read it with the caveats above
These are the headline numbers as reported by each model's authors. Treat each as a hypothesis, note the source type, and beat a trivial baseline before trusting any of them. The "Source type" column is doing real work here: a result in Science or Nature Methods has cleared independent review; a result in a release blog or preprint has not.
| Model | Task | Reported result | Source type |
|---|---|---|---|
| ESM-1v | Zero-shot VEP (~41 DMS sets) | Comparable to DeepSequence / EVmutation17 | NeurIPS 2021 |
| ESMFold | Structure (CAMEO) | TM-score ~0.71, 6x-60x faster3 | Science 2023 |
| ProGen | Lysozyme generation | 73% functional vs 59% natural5 | Nature Biotech 2023 |
| ESM3 | Novel GFP design | esmGFP, 58% identity to nearest1 | Science 2025 |
| PoET-2 | ProteinGym (<10 homologs) | Better perplexity, up to 80x smaller18 | arXiv preprint |
| AMPLIFY 350M | Sequence-only tasks | Matches/surpasses ESM2-15B8 | bioRxiv preprint |
| ESM C | Embedding efficiency | 600M rivals ESM2-3B4 | Release blog |
| ProstT5 | Remote homology (SCOPe40) | ROC-AUC ~0.45 vs ~0.06 seq-only12 | NAR Genom. Bioinf. 2024 |
| ProtT5-XL | Secondary structure Q3 | ~81-87%7 | IEEE TPAMI |
| Ankh3-XL | Secondary structure Q3 (CASP12) | up to 84.40%11 | README / papers |
| SaProt | 10 downstream tasks | Surpasses ESM-2, ESM-1v, MIF-ST13 | ICLR 2024 |
| xTrimoPGLM-100B | 18 understanding benchmarks | SOTA on most14 | Nature Methods 2025 |
The peer-reviewed entries carry more weight than the blog-and-preprint entries, not because the latter are wrong but because they have not yet survived independent review. The PoET-2 claim of beating much larger models "by a large margin" would matter a great deal if it holds; it is an arXiv preprint with vendor framing, so I would want a third party to reproduce the low-homolog result before betting a pipeline on it.18 The ProstT5 row is the opposite, a clean validity-aware claim: it states the baseline it beats and the cost it pays, ROC-AUC about 0.45 against sequence search at 0.06, at roughly a thousandth of the cost of full folding.12 That is the shape every benchmark claim should take.
The licensing fault line: the open era and the enclosure
The single most consequential thing about this field for a working lab is not architecture. It is the split that happened when the ESM team left Meta to found EvolutionaryScale. Read the dates and the licenses together and the story tells itself.
In 2019, Meta AI released ESM-1b, MIT, derived from training on about 250 million protein sequences clustered to UniRef50.2 In 2021, ESM-1v arrived, also MIT, a five-model ensemble trained on UniRef90 for zero-shot variant scoring.17 In 2022, ESM-2 scaled the recipe to 15B parameters and powered ESMFold, all MIT.3 The official facebookresearch/esm repository was archived read-only on August 1, 2024.3 By then the people had moved.
EvolutionaryScale, founded by former Meta FAIR ESM team members, released ESM3 in June 2024, peer-reviewed in Science in January 2025.1 It is a genuine advance over ESM-2 in scope, adding structure and function as fused tracks, and esmGFP is real.1 And the open weights, including the 1.4B ESM3-open model, are released under the Cambrian Non-Commercial License: research only, no commercial use, no providing outputs as a service, with a "Built with ESM" attribution requirement.1 The 7B and 98B models are API-only.1 Then ESM C followed in December 2024, tiered: the 300M model and all code are under the permissive Cambrian Open License, the 600M weights are non-commercial, and the 6B is API-only.4 The lineage that gave the field its open foundation now gates its frontier.
This is not a moral complaint. EvolutionaryScale is a company and the Cambrian terms are clear about what they permit. The practitioner point is narrower and more useful: ESM-2 did not get worse when ESM3 shipped. The capability frontier and the deployment frontier have separated, and you should track them separately. Here is where the whole field sits on the deployment ledger.
| Model | Code license | Weights license | Commercial use | Largest open |
|---|---|---|---|---|
| ESM-1b | MIT2 | MIT2 | Allowed2 | 650M2 |
| ESM-1v | MIT17 | MIT17 | Allowed17 | 650M x517 |
| ESM-2 | MIT3 | MIT3 | Allowed3 | 15B3 |
| ESM3 | Permissive1 | Cambrian Non-Commercial1 | Separate license needed1 | 1.4B1 |
| ESM C | Cambrian Open4 | 300M open / 600M NC / 6B API4 | Tier-dependent4 | 300M open4 |
| ProGen2 | BSD-36 | BSD-36 | Allowed6 | 6.4B6 |
| ProtGPT2 | Apache-2.09 | Apache-2.09 | Allowed9 | 738M9 |
| SaProt | MIT13 | MIT13 | Allowed13 | 1.3B13 |
| ProstT5 | MIT12 | MIT12 | Allowed12 | 3B12 |
| AMPLIFY | MIT8 | MIT8 | Allowed8 | 350M8 |
| CARP | BSD-style16 | BSD-style16 | Not prohibited16 | 640M16 |
| ProtBERT | AFL-3.07 | AFL-3.07 | Allowed7 | 420M7 |
| ProtT5 | MIT (code)7 | AFL-3.0 weights7 | Allowed7 | 11B7 |
| Ankh | CC-BY-NC-SA-4.011 | CC-BY-NC-SA-4.011 | Prohibited11 | ~2B11 |
| xTrimoPGLM | CC-BY-NC-4.014 | CC-BY-NC-4.014 | Prohibited14 | 100B INT414 |
| PoET / PoET-2 | MIT (code)15 | Non-Commercial15 | Prohibited15 | 182M18 |
| ProLLaMA | Llama-2 Community10 | Llama-2 Community10 | Restricted10 | ~7B10 |
Three traps in this table catch people in compliance review.
First, code license is not weights license. PoET's inference code is MIT, but its weights are non-commercial.15 ProtT5 ships MIT code and AFL-3.0 weights.7 You cannot read the GitHub badge and assume you are clear.
Second, the "Academic Free License" is permissive, not non-commercial. ProtBERT and ProtT5 weights are AFL-3.0, an OSI-approved license that does permit commercial use despite the misleading name.7 Conversely, Ankh's academic provenance does not save you: its license is CC-BY-NC-SA-4.0, genuinely non-commercial, requiring a separate agreement from Proteinea for any commercial deployment.11
Third, ProLLaMA's apache-2.0 tag is misleading. The model is built on LLaMA-2-7B, and both the card and repo instruct users to follow the Llama 2 license, so the binding terms are the Meta Llama 2 Community License.10
If you are building a product, the practical universe of fully open, commercially usable PLMs is smaller than the field looks: the ESM-2 line, ProGen, ProGen2, ProtGPT2, ProstT5, SaProt, AMPLIFY, CARP, ProtBERT, and ProtT5. Almost everything at the multimodal-generative frontier carries strings.
What is not known
Honesty about gaps is part of the job, so here is what this survey cannot tell you.
Several parameter counts are genuinely undisclosed or inconsistent. The original PoET checkpoint's exact size is unknown, documented only as a roughly 400 MB file.15 Ankh's large-variant counts differ depending on whether you count the encoder or the full encoder-decoder.11 ESM3's 7B and 98B models are API-only, so no one outside EvolutionaryScale can independently audit them.1 ProLLaMA's final trainable count beyond the 7B LLaMA-2 backbone is not separately disclosed, since it trains LoRA adapters.10
Independent evaluation is thin. Most benchmark numbers here are author-reported on author-chosen tasks. ESM C's efficiency claims have no peer-reviewed paper behind them yet.4 AMPLIFY's "is scaling necessary" thesis is a bioRxiv preprint, and some of its per-task tables could not be independently re-extracted.8 The vendor framing around PoET-2 is a marketing extrapolation, not an audited measurement.18 License terms also drift: ProLLaMA's superfamily exact-match rate moved from 62% to 67.1% across revisions, so cite the version you used.10
And the deepest unknown is generation hit rates. esmGFP is real,1 ProGen's lysozymes are real and wet-lab validated,5 but neither tells you the denominator: how many candidates you must synthesize and screen to get one that works for your target. Until generative PLMs report calibrated success rates rather than selected successes, treat de novo design as a candidate-enrichment tool, not a solved problem.
The honest recommendation
If you take one thing from both ledgers, take this: for the large majority of tasks a working bioinformatician faces in 2026, a well-tuned ESM-2 650M is still the right default. It is MIT-licensed,3 it runs on hardware you already have, it is the most-benchmarked model in the field, and the cases where a 100B model decisively beats it are narrower than the headlines suggest. AMPLIFY 350M and ProGen2 are the productivity wins. ESM-1v is the variant-scoring baseline you should beat before believing anything fancier.
Reach for the frontier when you have a specific reason: PoET-2 when homology is sparse and you can live with non-commercial terms,18 SaProt13 or ProstT512 when structure tokens demonstrably help your task, ESM3 when you genuinely need promptable multimodal generation and have a license arrangement.1 Everywhere else, the boring choice is the correct one.
Keep both ledgers open. A model that skips 500 million years of evolution on the capability ledger can still be the wrong line item on the validity ledger, and knowing the difference is most of what separates a useful pipeline from an impressive demo.
References
Footnotes
-
Hayes, T., Rao, R., Akin, H., Rives, A., et al., "Simulating 500 million years of evolution with a language model," Science (2025), DOI: 10.1126/science.ads0018. https://www.science.org/doi/10.1126/science.ads0018 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20 ↩21 ↩22 ↩23 ↩24 ↩25 ↩26 ↩27 ↩28
-
Rives et al., "Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences," PNAS 118(15):e2016239118, 2021. https://www.pnas.org/doi/10.1073/pnas.2016239118 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13
-
Lin et al., "Evolutionary-scale prediction of atomic-level protein structure with a language model," Science 379:1123-1130 (2023). https://www.science.org/doi/10.1126/science.ade2574 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20 ↩21 ↩22 ↩23 ↩24 ↩25 ↩26
-
"ESM Cambrian: Revealing the mysteries of proteins with unsupervised learning," EvolutionaryScale blog, Dec 4, 2024. https://www.evolutionaryscale.ai/blog/esm-cambrian ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19
-
Madani et al., "Large language models generate functional protein sequences across diverse families," Nature Biotechnology, 2023. https://www.nature.com/articles/s41587-022-01618-2 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10
-
Nijkamp, Ruffolo, Weinstein, Naik, Madani, "ProGen2: Exploring the Boundaries of Protein Language Models," arXiv:2206.13517 (2022); Cell Systems (2023). https://arxiv.org/abs/2206.13517 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12
-
Elnaggar et al., "ProtTrans: Towards Cracking the Language of Life's Code Through Self-Supervised Deep Learning and High Performance Computing," IEEE TPAMI (2022; arXiv:2007.06225, 2020). https://arxiv.org/abs/2007.06225 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20 ↩21
-
Fournier et al., "Protein Language Models: Is Scaling Necessary?" bioRxiv 2024. https://www.biorxiv.org/content/10.1101/2024.09.23.614603 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17
-
Ferruz, N., Schmidt, S. & Höcker, B., "ProtGPT2 is a deep unsupervised language model for protein design," Nature Communications 13, 4348 (2022). https://doi.org/10.1038/s41467-022-32007-7 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10
-
Lv et al., "ProLLaMA: A Protein Large Language Model for Multi-Task Protein Language Processing," arXiv:2402.16445 (2024; v3 2025). https://arxiv.org/abs/2402.16445 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13
-
Elnaggar et al., "Ankh: Optimized Protein Language Model Unlocks General-Purpose Modelling," bioRxiv 2023 / arXiv:2301.06568. https://arxiv.org/abs/2301.06568 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13
-
Heinzinger M. et al., "Bilingual language model for protein sequence and structure," NAR Genomics and Bioinformatics, 6(4):lqae150, 2024. https://doi.org/10.1093/nargab/lqae150 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14
-
Su et al., "SaProt: Protein Language Modeling with Structure-aware Vocabulary," bioRxiv 2023 / ICLR 2024. https://www.biorxiv.org/content/10.1101/2023.10.01.560349v1 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15
-
Chen et al., "xTrimoPGLM: unified 100-billion-parameter pretrained transformer for deciphering the language of proteins," Nature Methods (2025). https://www.nature.com/articles/s41592-025-02636-z ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15
-
Truong Jr & Bepler, "PoET: A generative model of protein families as sequences-of-sequences," NeurIPS 2023. https://arxiv.org/abs/2306.06156 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11
-
Yang, Fusi, Lu, "Convolutions are competitive with transformers for protein sequence pretraining," Cell Systems 15(3):286-294 (2024). https://www.cell.com/cell-systems/fulltext/S2405-4712(24)00029-2 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13
-
Meier, Rao, Verkuil, Liu, Sercu, Rives, "Language models enable zero-shot prediction of the effects of mutations on protein function," NeurIPS 2021; bioRxiv 2021.07.09.450648. https://www.biorxiv.org/content/10.1101/2021.07.09.450648v2 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14
-
Truong Jr & Bepler, "Understanding protein function with a multimodal retrieval-augmented foundation model" (PoET-2), arXiv:2508.04724 (2025). https://arxiv.org/abs/2508.04724 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11
Comments and feedback
Spotted an error or have a counterpoint? Comment below. No account needed, a name is enough. Corrections and pushback are welcome.