A 1-billion-parameter model conditioned on RNA chemical-probing reactivity folds a held-out transcript to an F1 of 0.987 against an experimentally guided reference. The same transcript folded from sequence alone scores 0.404.1 Across 1,000 held-out RNAs the improvement is robust, with a one-sided paired Wilcoxon signed-rank test at P = 6.25e-47.2 That is the cleanest single demonstration of what MIMIC is built to do: treat sequence, structure, regulation, evolutionary constraint, and experimental context as different observations of one underlying molecular state, then fill in whichever observation you withhold.
MIMIC, from Golkar et al. at Polymathic AI, is a generative multimodal foundation model posted to arXiv on 27 April 2026.34 It trains on a new aligned dataset called LORE, which links nucleic acid, protein, evolutionary, structural, regulatory, and semantic modalities within partially observed biomolecular states.3 A split-track encoder-decoder conditions on arbitrary subsets of observed modalities and reconstructs or generates the missing components across genome, transcriptome, and proteome.5 The headline is not the parameter count. The authors state the thesis plainly: at a comparable size to single-modality foundation models, around 1 billion parameters, MIMIC demonstrates the value of diverse and heterogeneous data as an alternative to pure scaling.6 If that holds, it is a different growth axis than the sequence-token-count race that produced Evo 2 and the ESM line.
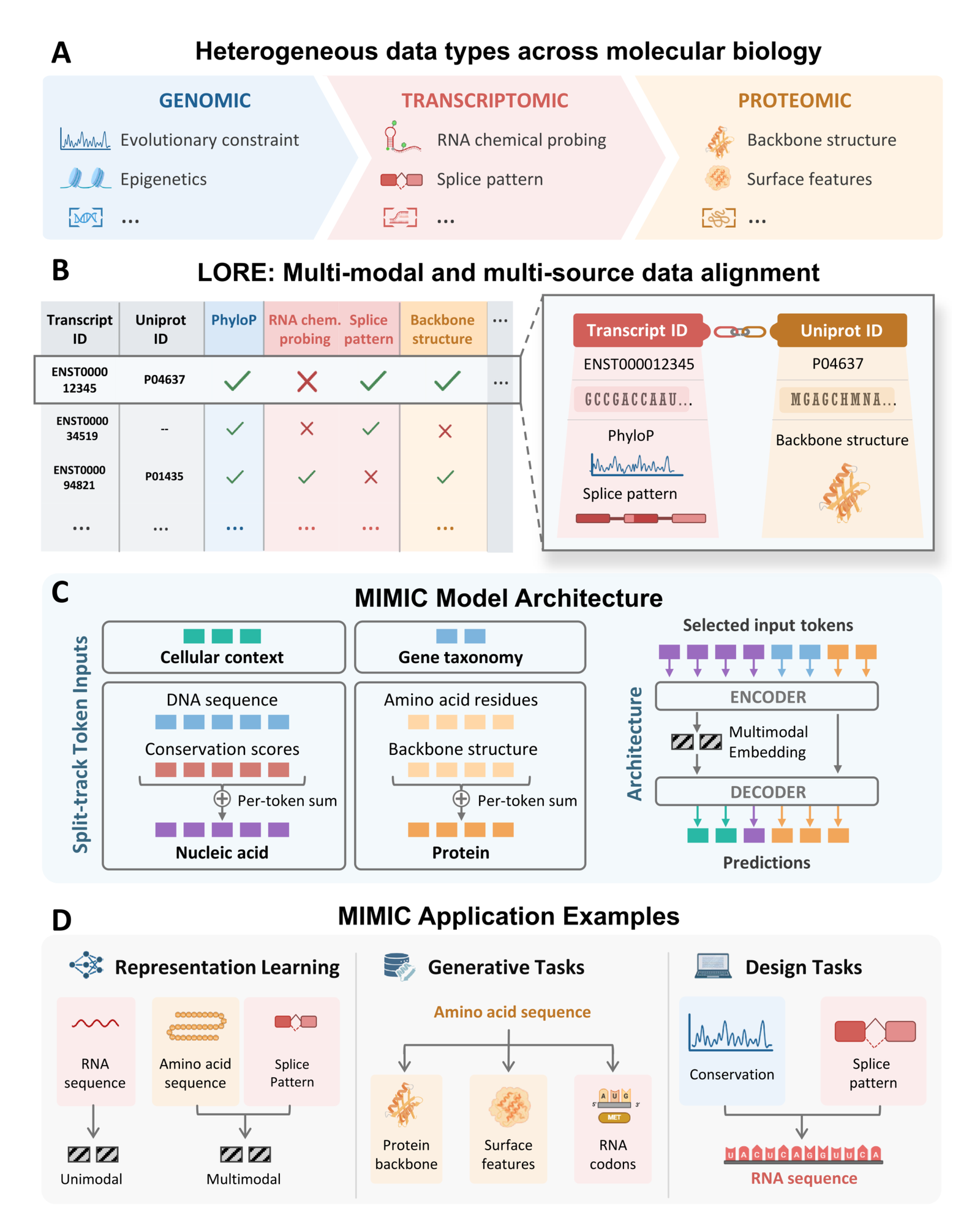 Figure 1: The MIMIC framework. (A) heterogeneous molecular data spanning genomic, transcriptomic, and proteomic modalities; (B) the LORE dataset aligning these into partially observed examples; (C) the split-track encoder-decoder; (D) representation, generative, and design applications. Source: Golkar et al., MIMIC (arXiv:2604.24506, 2026).
Figure 1: The MIMIC framework. (A) heterogeneous molecular data spanning genomic, transcriptomic, and proteomic modalities; (B) the LORE dataset aligning these into partially observed examples; (C) the split-track encoder-decoder; (D) representation, generative, and design applications. Source: Golkar et al., MIMIC (arXiv:2604.24506, 2026).
The problem: forward predictors model the wrong distribution
You already know the zoo. ESM and ESM-2 for protein sequence, RNA-FM for non-coding RNA, SpliceAI for splice sites, AlphaGenome for regulatory genomics, AlphaFold for structure. Each is excellent inside its lane. The field has accumulated, in the authors' words, single-modality models that master one molecular modality but lack the capacity to operate across the broader central dogma.7
MIMIC's critique is sharper than "models are siloed." It is that forward predictors model the wrong object. By mapping a sequence to a specific phenotype, these models fail to capture the full joint distribution of biological states; to understand a system, the argument goes, you model the full joint probability distribution of its molecular states, not just the forward flow of information.89 That reframing is what makes inverse problems tractable. Given a desired protein structure, mRNA stability, and splicing pattern, what upstream nucleotide sequence most likely produced them?10 A forward predictor cannot answer that; a generative model over the joint distribution can, by sampling the conditional.
This is a different bet than AlphaFold3, which is also a unified framework but a forward, structure-focused one: it predicts the joint structure of complexes including proteins, nucleic acids, small molecules, ions, and modified residues.11 AlphaFold3 unifies outputs, the structures of complexes. MIMIC unifies both inputs and outputs across the central dogma, and it is generative over sequence as well as structure. It also departs from the sequence-only RNA and protein language models it competes with. RNA-FM, for instance, is a 12-layer transformer trained on 23 million non-coding RNA sequences through self-supervised learning.12 MIMIC's question is what happens when you stop treating non-sequence signals as downstream tasks and start treating them as conditioning inputs.
LORE: the dataset is the contribution
The data is the part that took the most work. LORE holds roughly 15.5 million proteins and 13 million RNA transcripts from over 6,000 organisms spanning all domains of life, plus over 4 billion tokens of biomedical and contextual text.13 Those are post-clustering numbers. Before deduplication, the nucleic-acid side spans over 25 million transcripts and 123 billion nucleotides, from full human and mouse genomes with GENCODE annotations plus NCBI RefSeq for the rest.1415
The central design principle is alignment, not volume, which distinguishes LORE from sequence-only corpora such as OpenGenome2 or BFD that do not provide aligned or multimodal samples.16 Each row is an observation of a transcript, a protein, or a pairing of the two, containing whatever subset of modalities is available; mapping UniProt IDs to RefSeq/Ensembl IDs yields a core subset of roughly 2 million aligned transcript/protein pairs.1718 Because real data is incomplete, LORE is intentionally partially observed: no example is required to contain all modalities, which across the dataset produces about 150 distinct modality presence signatures.1920 That partial observation is not a flaw worked around later; it is the training signal, and it dictates the architecture below.
The per-modality counts make the raggedness concrete.
| Track | Modality | Samples in LORE |
|---|---|---|
| Nucleic acid | Nucleic acid sequence | 12,967,15321 |
| Nucleic acid | Promoter usage (CAGE) | 15,073,29822 |
| Nucleic acid | Chromatin accessibility (ATAC-seq) | 14,021,30122 |
| Nucleic acid | RNA chemical probing (RASP2) | 1,644,40423 |
| Nucleic acid | Evolutionary conservation (phyloP) | 518,48024 |
| Protein | AA sequence / DSSP / backbone / SASA | 15,607,838 each25 |
| Protein | Chemical surface (MaSIF) | 1,671,20626 |
| Protein | Protein abundance | 1,803,02826 |
| Protein | Functional captions | 176,62626 |
| Semantic | Biomedical corpus | 3,797,568 (4B tokens)27 |
Two orders of magnitude separate the common modalities (sequence, amino acids, epigenetic peaks) from the scarce ones (phyloP, MaSIF, functional captions). A model requiring all modalities present would train on the intersection, which is nearly empty. Two provenance details matter downstream. phyloP conservation comes from 100-way alignments for human and 30-way for mouse, capturing lineage-specific constraint.28 MaSIF surface fingerprints were computed on only about 10% of structures because of compute cost, roughly 50,000 CPU hours for 1.6M datasets. That is why the surface modality is two orders of magnitude smaller than the protein sequence count.29
Leakage control is handled with MMseqs2 clustering, run with -c 0.8 and --min-seq-id 0.3 or 0.7.30 Protein clustering at 30% identity collapses 299,566,796 members into 42,943,944 clusters; RNA collapses 25,001,445 into 10,861,904.31 Splits are made at the cluster level so no validation sequence shares significant similarity with training, with 30% clusters defining train/validation/test splits and 70% clusters ensuring diversity.32 For computational biologists, that is the line that determines whether the benchmark numbers below are believable, and it is the right line to draw. Tertiary structure is tokenized with the ESM3 structure tokenizer, a VQ-VAE with 4,096 classes based on local backbone geometry, applied to roughly 200M AlphaFoldDB v4 structures filtered to pLDDT greater than 70.3334
Architecture: split-track summation and an asymmetric window
The core engineering problem is that biological modalities have wildly different lengths and alignment structures: a single-value abundance measurement, a 10,000-nucleotide transcript, and a backbone track for a 400-residue protein all coexist in one model. MIMIC's answer is a split-track architecture, built on the 4M/AION encoder-decoder backbone, that sums aligned modalities while concatenating distinct molecular entities.3536
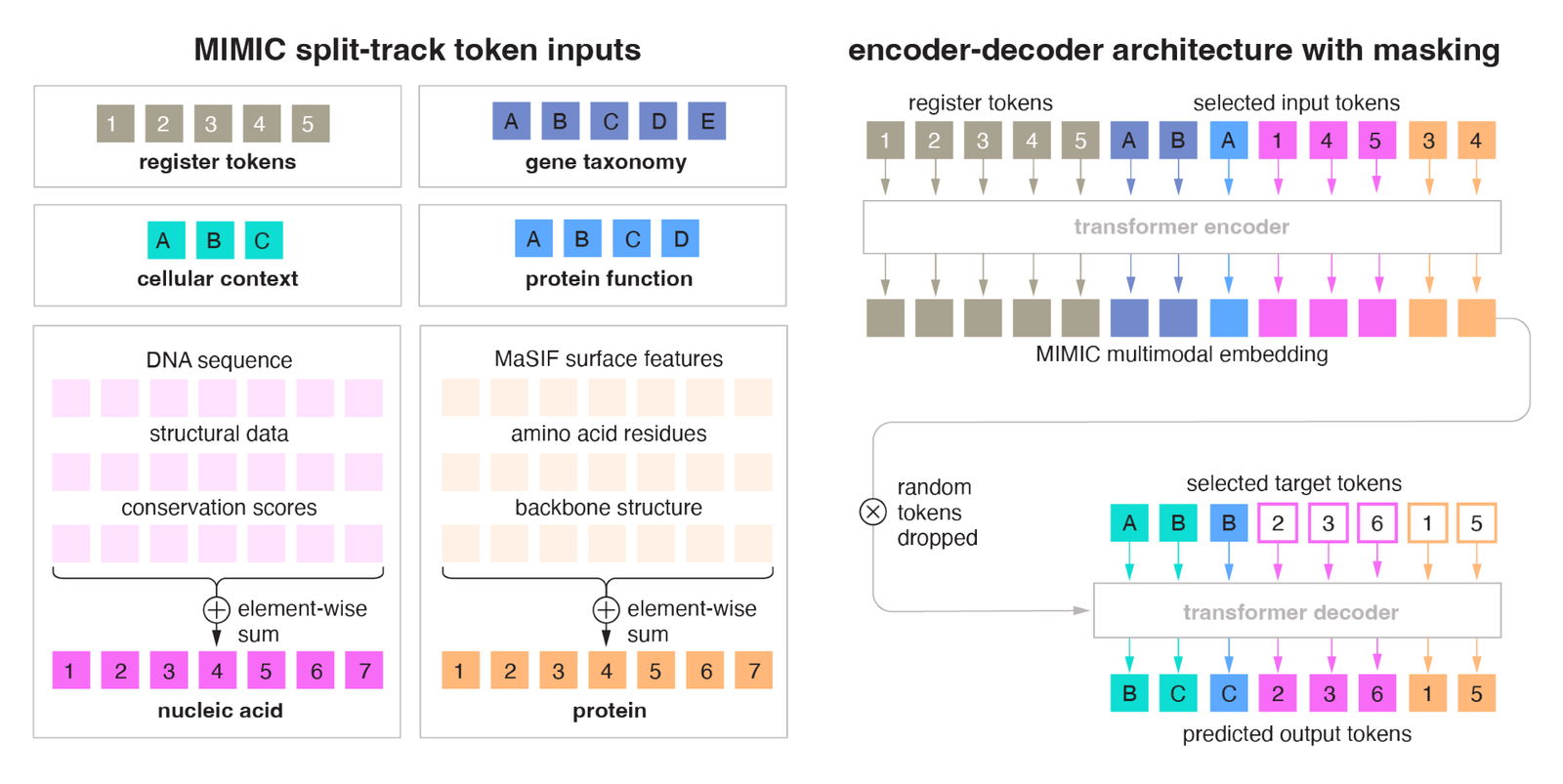 Figure 2: MIMIC's split-track architecture. Left: token inputs grouped into a nucleic-acid track (sequence + structural data + conservation, element-wise summed), a protein track (MaSIF surface + amino acids + backbone, element-wise summed), plus register, taxonomy, and context tokens. Right: the masked encoder-decoder. Source: Golkar et al., MIMIC (arXiv:2604.24506, 2026), Supplementary Figure S1.
Figure 2: MIMIC's split-track architecture. Left: token inputs grouped into a nucleic-acid track (sequence + structural data + conservation, element-wise summed), a protein track (MaSIF surface + amino acids + backbone, element-wise summed), plus register, taxonomy, and context tokens. Right: the masked encoder-decoder. Source: Golkar et al., MIMIC (arXiv:2604.24506, 2026), Supplementary Figure S1.
The nucleic-acid track element-wise sums sequence embeddings, structural data, and conservation scores into one representation per position; the protein track does the same for surface features, amino-acid residues, and backbone structure.37 Because aligned signals are summed rather than concatenated, total length stays tractable no matter how many modalities are present. A per-position phyloP score and the nucleotide at that position occupy the same token slot rather than doubling the sequence.38
The encoder processes these track groups alongside semantic and contextual token groups and a set of learnable register tokens, which act as information sinks pooling global state into a compact vector summary.3940 All text-based modalities (context, functional captions, taxonomy) share one semantic embedding space with the biomedical corpus.41 Positional encoding uses RoPE with a local group-reset strategy: the position index restarts at zero for each track group, so attention reflects within-track distances rather than absolute offsets. That is the detail that makes summing two differently-scaled tracks coherent.42
The decoupling of observed context from predicted output is the design's center of gravity. MIMIC uses an asymmetric context window, 10,000 tokens for the encoder and 1,000 for the decoder.43 The wide encoder receptive field ingests arbitrary observed context; the focused 1,000-token cross-attention decoder queries the encoder's latent to reconstruct or generate any target subset, with no architectural modification or task-specific output heads.44 That makes "condition on any subset, generate any subset" a single forward pass rather than a family of fine-tuned models. The cross-attention bottleneck descends from Perceiver IO, which replaces the usual modality-specific encoder plus fusion plus readout recipe with a flexible querying mechanism.45
The full configuration: 20 encoder layers, 12 decoder layers, hidden width 1,536, 24 attention heads per stack, 5 register tokens, RoPE fraction 0.75.46 Self-attention uses a 50% bidirectional, 25% causal, 25% anti-causal mask, following the finding that causal attention can outperform bidirectional on certain tasks.47 Training ran by default across 92 NVIDIA H200 GPUs over NCCL, using AdamW (betas 0.9/0.95, weight decay 0.05), cosine decay, and bfloat16.4849 Only the 566M-parameter encoder generates embeddings for downstream tasks, the relevant size when comparing representation quality against other models.50
Training: 25 pathways, a curriculum, and crop-versus-drop
There is no monolithic loss. Training is a stochastic sampling process over roughly 25 heuristic pathways, each specifying a required and optional set of input and target modalities.51 This is how MIMIC handles about 150 presence signatures without enumerating them: on each step a pathway is sampled, inputs are chosen from available modalities, and the model reconstructs the targets. The same machinery yields "predict masked sequence from sequence plus conservation" and "generate amino-acid sequence from backbone plus surface" without separate heads.
Three engineering choices encode real biology.
- Context curriculum. A staged schedule scales the encoder window from 1k to 2k to 4k to 8k to 10k tokens, so the model learns local feature associations before resolving long-range regulatory and structural dependencies.52
- Crop versus drop. RNA and DNA sequences are randomly cropped to fit the current window, which stays valid because RNA folding is largely driven by local base pairing. Proteins are dropped, not cropped: a truncated protein cannot reliably predict structure because long-range contacts go missing, so any protein whose structure track exceeds the budget is discarded entirely.5354
- Target packing. Many biological targets are sparse or short (splice-junction tracks, single-value abundance), so the decoder packs multiple into its 1,000-token budget.55 This acts as a regularizer, forcing the model to learn joint dependencies between, say, splicing and conservation even when the explicit task was only splicing.56
Register tokens are trained via reconstruction with 0 to 10% random token dropout across all input tracks, which prevents them from degenerating into trivial copy mechanisms.57 Mixing short peptides with long RNAs in one batch is also a real systems problem: naive padding wastes compute on heavy-tailed length distributions and naive packing truncates, so MIMIC uses dynamic workload balancing that routes short sequences into large batches and long sequences into small ones across GPUs.58
Reconstruction: a 1B model holding its own against larger ones
The headline reconstruction result uses a sequence-completion (inpainting) task. A 100-token window in the center of a sequence is masked and reconstructed in a single pass by sampling from a near-zero-temperature softmax over the model's logits.59 Random per-token accuracy is about 0.25 for nucleotides and about 0.04 for amino acids, which sets the floor.60
For protein sequence completion, MIMIC conditioned with structural descriptors reaches the highest reconstruction accuracy against ProtBERT, ESM-2, ESM-C, and ESM3-open.61 On unspliced transcripts, with masking stratified into exonic and intronic regions, MIMIC with the full set of aligned genomic modalities beats two long-context genomic models trained on OpenGenome2 in both regions: NTv3 (masked reconstruction) and Evo 2 (autoregressive).62 Even sequence-only it is competitive with the top sequence-only models, and conditioning on more modalities yields consistent gains.63 Beating a much larger autoregressive genomic model with a 1B model is the cleanest support for the diversity-over-scale thesis.
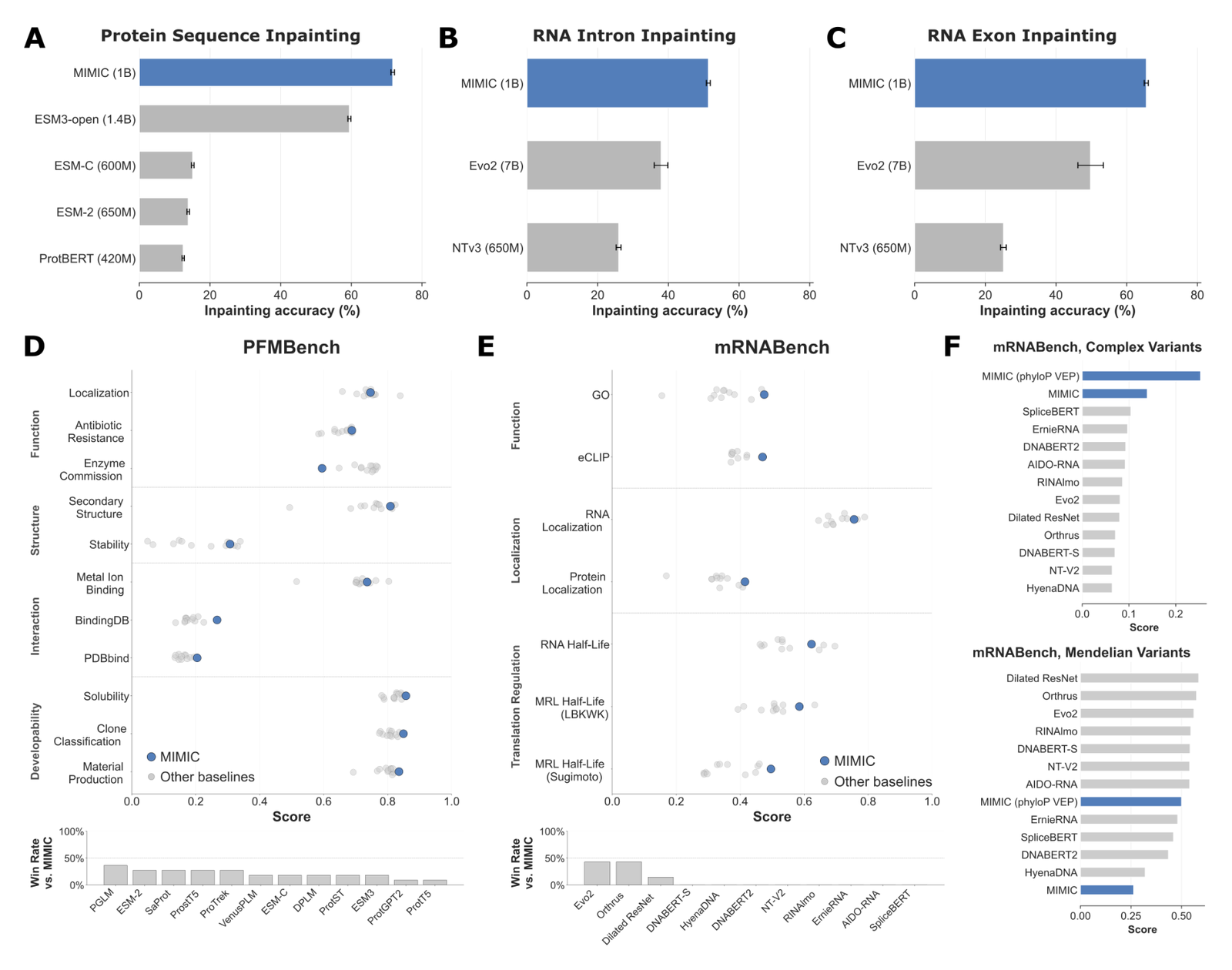 Figure 3: State-of-the-art sequence property prediction. (A) protein amino-acid inpainting: MIMIC vs ESM3-open, ESM-C, ESM-2 (650M), ProtBERT. (B-C) RNA intron/exon inpainting vs Evo 2 (7B) and NTv3 (650M). (D-E) PFMBench and mRNABench. (F) complex and Mendelian variant scores. Baseline names follow the paper's figure; the parenthetical parameter counts are each model's own published size. Source: Golkar et al., MIMIC (arXiv:2604.24506, 2026).
Figure 3: State-of-the-art sequence property prediction. (A) protein amino-acid inpainting: MIMIC vs ESM3-open, ESM-C, ESM-2 (650M), ProtBERT. (B-C) RNA intron/exon inpainting vs Evo 2 (7B) and NTv3 (650M). (D-E) PFMBench and mRNABench. (F) complex and Mendelian variant scores. Baseline names follow the paper's figure; the parenthetical parameter counts are each model's own published size. Source: Golkar et al., MIMIC (arXiv:2604.24506, 2026).
Representation quality holds up on standard probes. On PFMBench (protein function, structure, interaction, and developability across 11 tasks), MIMIC matches or exceeds ESM3, ESM-C, ProTrek, and SaProt, winning at least 7 of 11 tasks against each competitor.64 It is strongest on protein-ligand binding (BindingDB, PDBbind) and best on every developability task, which the authors attribute to jointly pretraining on surface chemistry.65
On mRNABench, MIMIC beats Evo 2 and Orthrus on 4 of 7 tasks and a Dilated ResNet on 6 of 7. Its largest gains are on aligned-modality tasks: GO function prediction (+2% vs Evo 2, +9% vs Orthrus) and protein localization (+2% vs Evo 2, +5% vs Orthrus).6667 The honest negatives sit in the same table: MIMIC is 8% ahead of Orthrus on the Sugimoto half-life set but 8% behind on the LBKWK half-life set, a benchmark from synthetic mRNA constructs where stability effects likely dominate.6869 All of this comes from the 566M encoder, one of the smaller networks in the comparison.50
Ablations: does multimodality actually help, and when?
The thesis is that diverse data beats scaling, so the ablations are the load-bearing evidence. A skeptic's first question is whether the extra modalities do real work or just add parameters. They do real work, but unevenly.
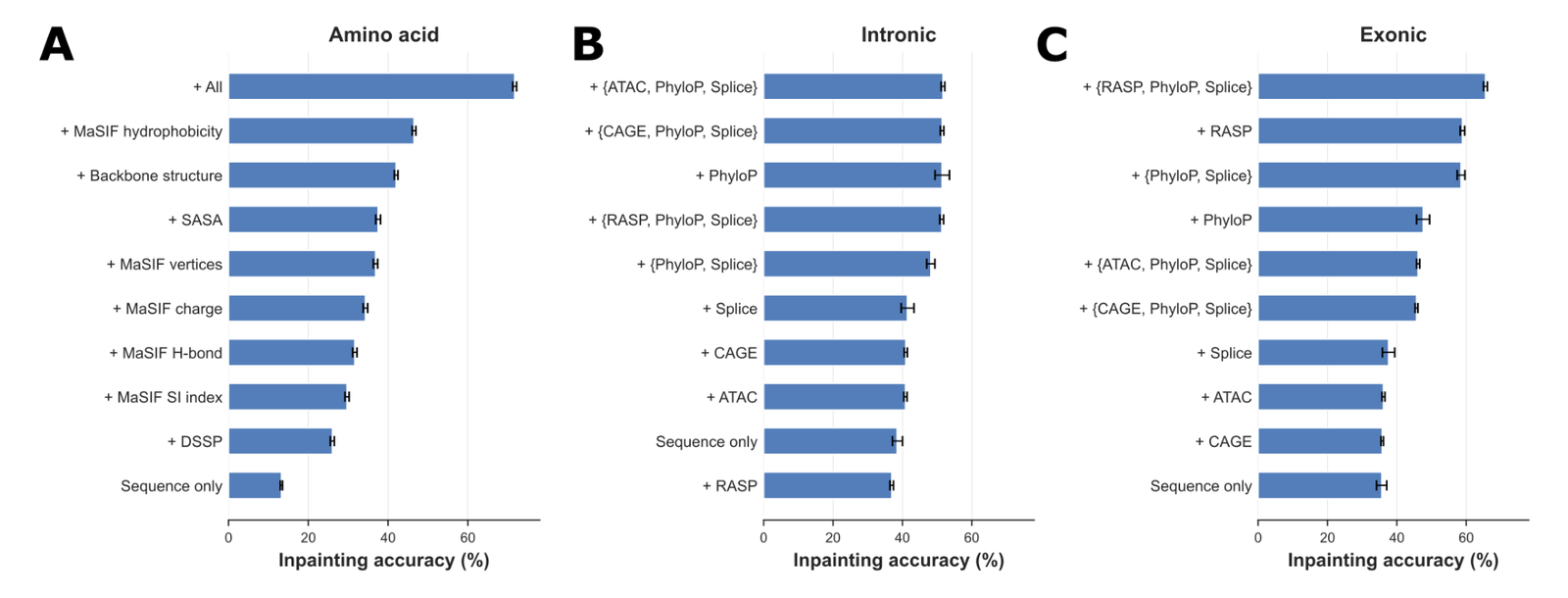 Figure 4: Ablation on conditioning inputs, sorted by inpainting accuracy. (A) amino acids: sequence-only rising to all structural/chemical modalities. (B) intronic and (C) exonic RNA: effect of adding ATAC, phyloP, splice, CAGE, RASP and combinations. Source: Golkar et al., MIMIC (arXiv:2604.24506, 2026), Supplementary Figure S3.
Figure 4: Ablation on conditioning inputs, sorted by inpainting accuracy. (A) amino acids: sequence-only rising to all structural/chemical modalities. (B) intronic and (C) exonic RNA: effect of adding ATAC, phyloP, splice, CAGE, RASP and combinations. Source: Golkar et al., MIMIC (arXiv:2604.24506, 2026), Supplementary Figure S3.
For amino acids, the most effective single modalities are MaSIF hydrophobicity and tokenized backbone structure; for intronic sequence, evolutionary information (phyloP), splicing, and epigenetics (ATAC-seq or CAGE) drive the gains. In both cases combining all of them beats any single addition.7071 The cross-cutting rule, and the one that protects the work from hype: multimodal conditioning is most useful where the auxiliary modality is ambiguous from sequence alone, rather than easily predictable from it.72
That point is sharpened in a separate analysis that bins the uplift by how predictable each modality is from sequence.
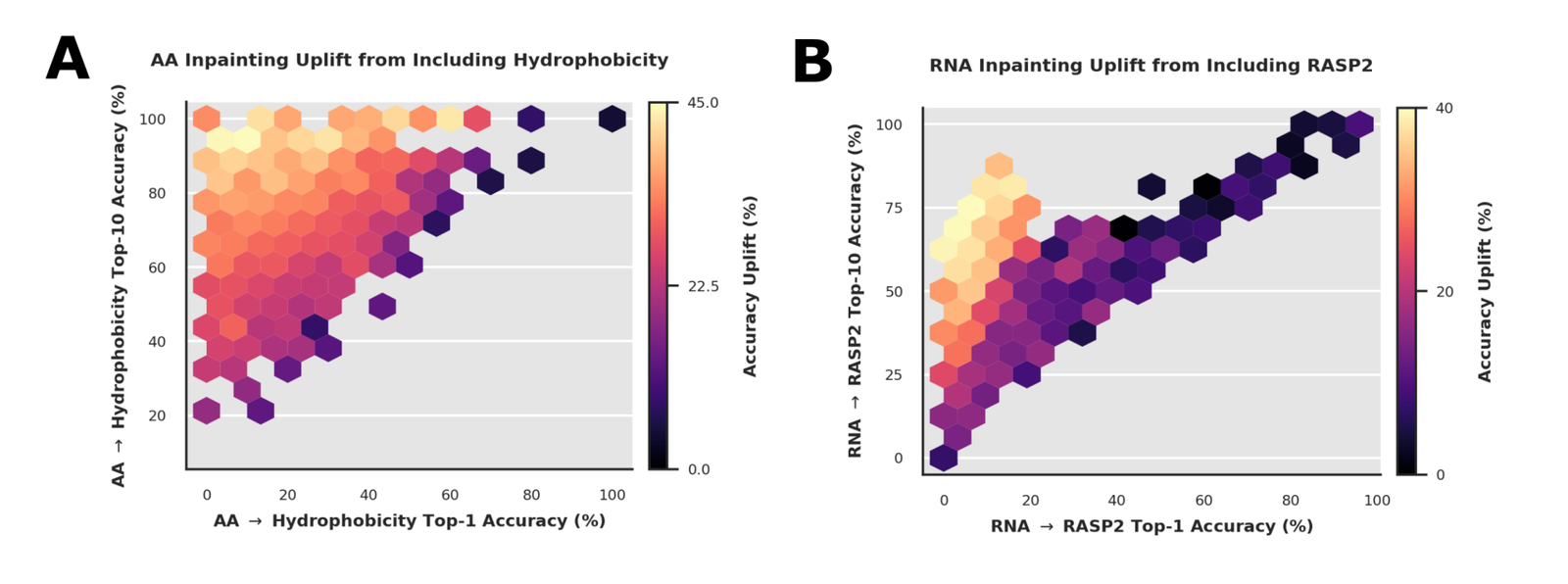 Figure 5: Performance gains depend on modality predictability. (A) amino-acid inpainting uplift from hydrophobicity, binned by how well hydrophobicity is predicted from sequence. (B) RNA inpainting uplift from RASP2. Uplift peaks when a modality is informative but not trivially predictable from sequence. Source: Golkar et al., MIMIC (arXiv:2604.24506, 2026), Supplementary Figure S4.
Figure 5: Performance gains depend on modality predictability. (A) amino-acid inpainting uplift from hydrophobicity, binned by how well hydrophobicity is predicted from sequence. (B) RNA inpainting uplift from RASP2. Uplift peaks when a modality is informative but not trivially predictable from sequence. Source: Golkar et al., MIMIC (arXiv:2604.24506, 2026), Supplementary Figure S4.
This is the kind of result that earns credibility rather than spends it. If sequence already determines the modality, adding it is redundant; if the modality is noise, it adds nothing. The value lives in between, the expected signature of genuine information transfer. The scarce modalities in LORE, phyloP and MaSIF, are also the ones least predictable from sequence, which is exactly where the data table and the ablation meet: the rarest tracks carry the most non-redundant signal.
Splicing: matching the dedicated predictors
Splice-site detection is the cleanest head-to-head against specialized tools. Because splice sites are rare, with about 99.85% of nucleotide positions not splice sites, the metric is AUPR.73 To compare fairly, each model gets the context its design needs: SpliceAI receives 5kb flanks each side, AlphaGenome a 16,384 bp interval, MIMIC the full sequence up to 10kb.74 MIMIC's splice head outputs a 5-class distribution over non-splice, donor, acceptor, TSS, and TES at each position.75 Because many genes exceed the context length, evaluation is restricted to genes under 25kb, with genes over 10kb scored on a common 10kb region carrying the most junctions.76
| Level | Region | vs AlphaGenome | vs SpliceAI | vs NTv3 |
|---|---|---|---|---|
| Gene | coding | +1% | +3% | +11% |
| Gene | non-coding | +12% | +14% | +311% |
| Transcript | coding | +6% | +5% | +11% |
| Transcript | non-coding | +26% | +16% | +146% |
Splice-site AUPR deltas, MIMIC versus baselines. Source: Golkar et al., MIMIC (arXiv:2604.24506, 2026), Figure 3a-b.7778
At gene level MIMIC edges ahead of the dedicated forward predictors; at transcript level, where the joint generative formulation enables isoform-aware inference, it is clearly the strongest model.7778 One caveat on reading the table: the large non-coding multiples versus NTv3 (+311% and +146%) are gains over a weaker baseline on a rare-event metric, so they look more dramatic than the coding-region deltas against tuned splice predictors. Feeding the transcript start and end sites (TSS/TES) as explicit input improves recovery of internal splice structure by +3% coding and +5% non-coding, consistent with evidence that start-site usage shapes isoform selection.79
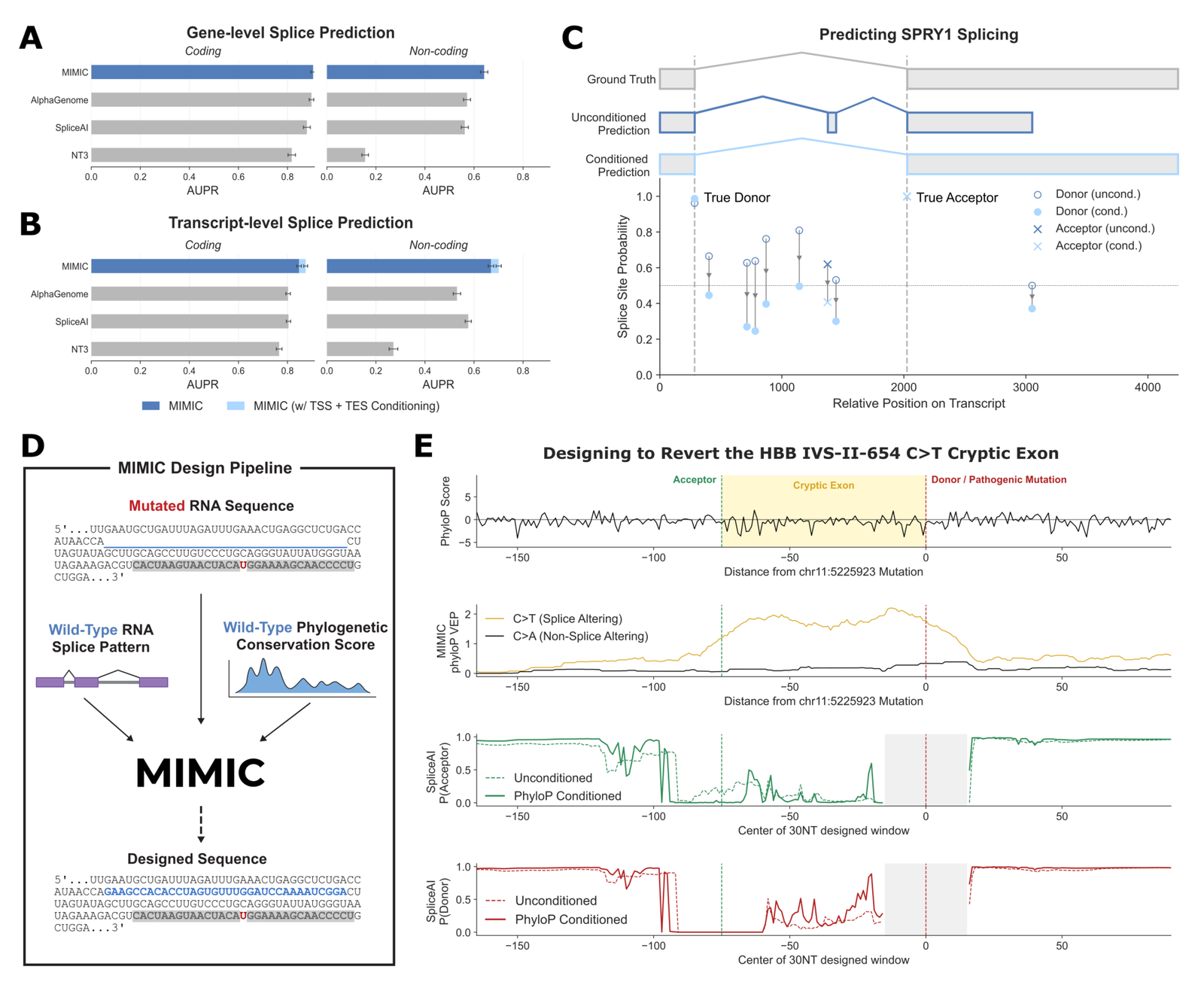 Figure 6: Splice-site prediction and RNA design. (A) gene-level AUPR (coding/non-coding) vs AlphaGenome, SpliceAI, NTv3. (B) transcript-conditioned AUPR. (C) SPRY1 prediction, unconditioned vs conditioned. (D-E) designing to revert the HBB IVS-II-654 C>T cryptic exon. Source: Golkar et al., MIMIC (arXiv:2604.24506, 2026).
Figure 6: Splice-site prediction and RNA design. (A) gene-level AUPR (coding/non-coding) vs AlphaGenome, SpliceAI, NTv3. (B) transcript-conditioned AUPR. (C) SPRY1 prediction, unconditioned vs conditioned. (D-E) designing to revert the HBB IVS-II-654 C>T cryptic exon. Source: Golkar et al., MIMIC (arXiv:2604.24506, 2026).
The SPRY1 example shows the mechanism. Before conditioning, MIMIC predicts an extra exon not in the ground truth plus several spurious donor sites; after conditioning on transcript boundaries, all false-positive junctions drop to low probability and only the true exons survive.80 The constraint is doing real work, not just rescoring.
Corrective RNA edits without reverting the mutation
The clinical demonstration targets the intronic beta-globin variant HBB c.316-197C>T (IVS-II-654 C>T), a canonical splice-altering mutation that promotes pseudoexon inclusion through aberrant splice-site gain.81 The constraint that makes it hard is fixing the splicing defect without reverting the causal mutation. That is the regime for RNA-level therapeutics, where A-to-I editing by ADAR enzymes can alter splice sites without touching the genome.82
MIMIC's phyloP variant-effect score distinguishes the pathogenic C>T from a matched non-splice-altering C>A control, localizing signal to the cryptic acceptor and donor.83 The design task prompts MIMIC with both the wild-type splice pattern and the pathogenic sequence with the C>T held fixed, generating edits in 30- or 50-nucleotide windows at varying distances. SpliceAI serves as an independent oracle, since scoring MIMIC's designs with MIMIC would be circular.8485 MIMIC produces designs that substantially reduce pathogenic splicing while keeping the causal mutation intact.86 The result that matters most: effective corrections appear even when the editable window does not overlap the cryptic splice sites, so the model is doing indirect regulatory rewiring through exonic splicing regulatory elements rather than just patching the obvious site.87
Protein binder design
For binder design the targets are PD-L1 (PDB 4ZQK chain A) and the larger hACE2 (6VW1 chain A), the latter against the SARS-CoV-2 spike RBD.88 The team generated sequence ensembles under five conditioning strategies, 20 per strategy: MaSIF surface features alone (40% or 100% of residues near the binding site), backbone geometry alone, or backbone plus partial or full surface.89 Generation is iterative, up to 10 cycles, with MIMIC used for both generation and verification; the top candidates are then folded with AlphaFold2, keeping designs at average pLDDT above 85 and scoring fidelity by TM-score against native.90
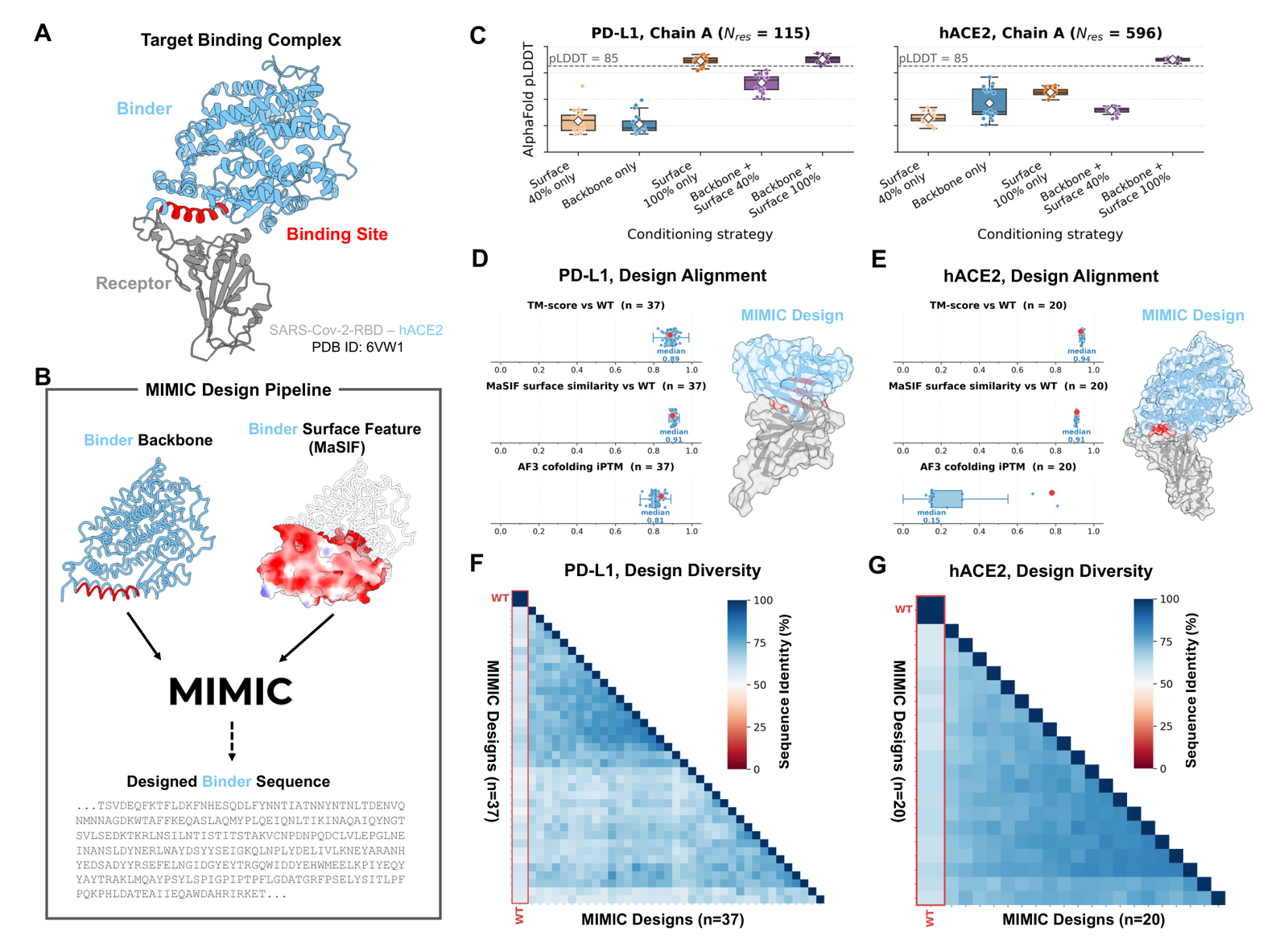 Figure 7: Target-binder design. (A) the target complex (SARS-CoV-2-RBD and hACE2, PDB 6VW1). (C) pLDDT across conditioning strategies. (D-E) TM-score, MaSIF similarity, and AlphaFold3 cofolding iPTM. (F-G) sequence-identity diversity heatmaps (PD-L1 n=37, hACE2 n=20). Source: Golkar et al., MIMIC (arXiv:2604.24506, 2026).
Figure 7: Target-binder design. (A) the target complex (SARS-CoV-2-RBD and hACE2, PDB 6VW1). (C) pLDDT across conditioning strategies. (D-E) TM-score, MaSIF similarity, and AlphaFold3 cofolding iPTM. (F-G) sequence-identity diversity heatmaps (PD-L1 n=37, hACE2 n=20). Source: Golkar et al., MIMIC (arXiv:2604.24506, 2026).
Design confidence peaked when both backbone and surface chemistry were provided, and the benefit scaled with target complexity: surface chemistry alone reached pLDDT >= 85 for the smaller PD-L1, while the larger hACE2 leaned much harder on added backbone conditioning.91 Designed sequences recovered the wild-type fold (median TM-score > 0.85) and surface chemistry (median MaSIF similarity > 0.90).92 Binding evidence comes from a separate step, in silico cofolding with AlphaFold3: for PD-L1, 35 of 37 high-confidence designs hit iPTM > 0.75 (median 0.81); for the harder hACE2, only 2 of 20 high-fold designs were predicted to bind the spike RBD.9394 Two tools do two jobs here: AlphaFold2 for fold confidence and TM-score, AlphaFold3 for cofolding iPTM. Diversity matrices show designs at only about 50% sequence identity to wild type, so this is not memorization, and the all-in-silico evidence is exactly why the independent oracle and cofolding checks matter.9596
RNA chemical probing as semantic conditioning
The most conceptually interesting result treats experimental context as a modality. Rather than fixed per-assay output heads, MIMIC processes natural-language descriptions of experimental conditions alongside molecular data, using context as semantic conditioning for assay-dependent chemical probing.97
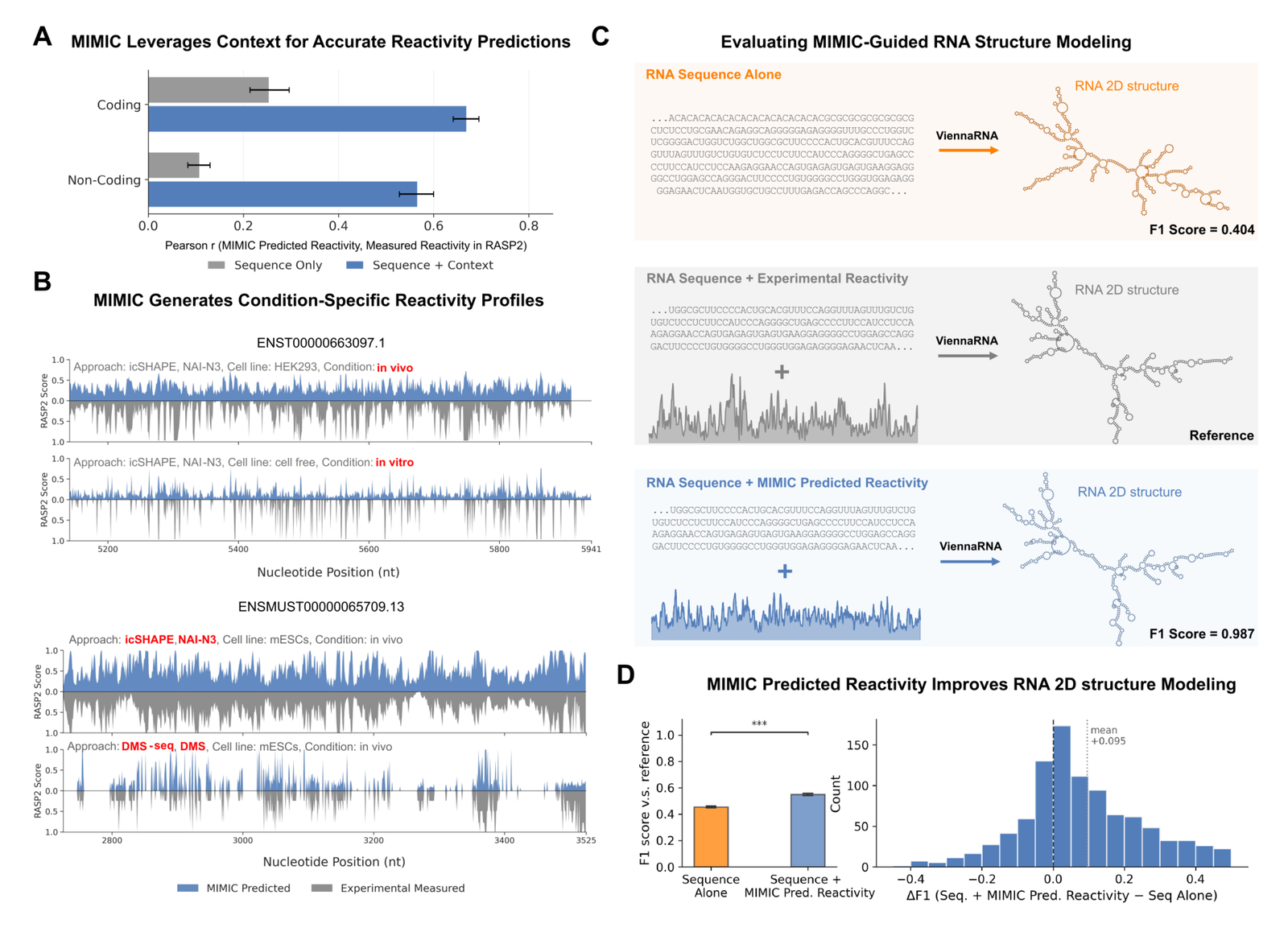 Figure 8: RNA chemical-probing reactivity and structure. (A) Pearson r for predicted vs measured RASP2, sequence-only vs sequence+context. (B) condition-specific reactivity profiles. (C) MIMIC-guided 2D structure: sequence alone (F1=0.404) vs MIMIC reactivity (F1=0.987) vs reference. (D) distribution of F1 improvement. Source: Golkar et al., MIMIC (arXiv:2604.24506, 2026).
Figure 8: RNA chemical-probing reactivity and structure. (A) Pearson r for predicted vs measured RASP2, sequence-only vs sequence+context. (B) condition-specific reactivity profiles. (C) MIMIC-guided 2D structure: sequence alone (F1=0.404) vs MIMIC reactivity (F1=0.987) vs reference. (D) distribution of F1 improvement. Source: Golkar et al., MIMIC (arXiv:2604.24506, 2026).
Conditioning on the correct experimental context significantly improves transcriptome-wide RASP2 reactivity prediction over sequence-only models.98 MIMIC distinguishes assay chemistry, separating the broad sensitivity of in vivo icSHAPE (NAI-N3 probe) from the A/C-restricted sensitivity of DMS-seq.99 Feeding predicted reactivity into ViennaRNA takes the illustrated example from F1 = 0.404 (sequence alone) to F1 = 0.987 (MIMIC reactivity), a structure nearly identical to the experimentally guided reference.100 Across 1,000 held-out transcripts, with three RNAfold (v2.7.2) predictions each, the improvement is robust: median delta F1 = +0.061, mean +0.095 +/- 0.194 s.d., with a one-sided paired Wilcoxon signed-rank test at P = 6.25e-47.101102 That Wilcoxon result is a statistically significant difference favoring MIMIC, not statistical equivalence; "nearly identical" describes the single illustrated transcript, not the whole distribution. Conditioning on phyloP conservation also lifts RASP2 accuracy, a structure-conservation synergy the authors liken to MSA coevolution in protein folding.103
Variant effect: generation beats embeddings
The variant-effect work shows a related subtlety: how you read MIMIC matters as much as what it knows. MIMIC can expose task-relevant signal directly through generation rather than forcing every task through a generic embedding.104 Its phyloP-based VEP scores a variant by the mean absolute perturbation in a 30-nucleotide window around the position, a low-dimensional regressor that is more interpretable and performs substantially better than embedding-based alternatives.105
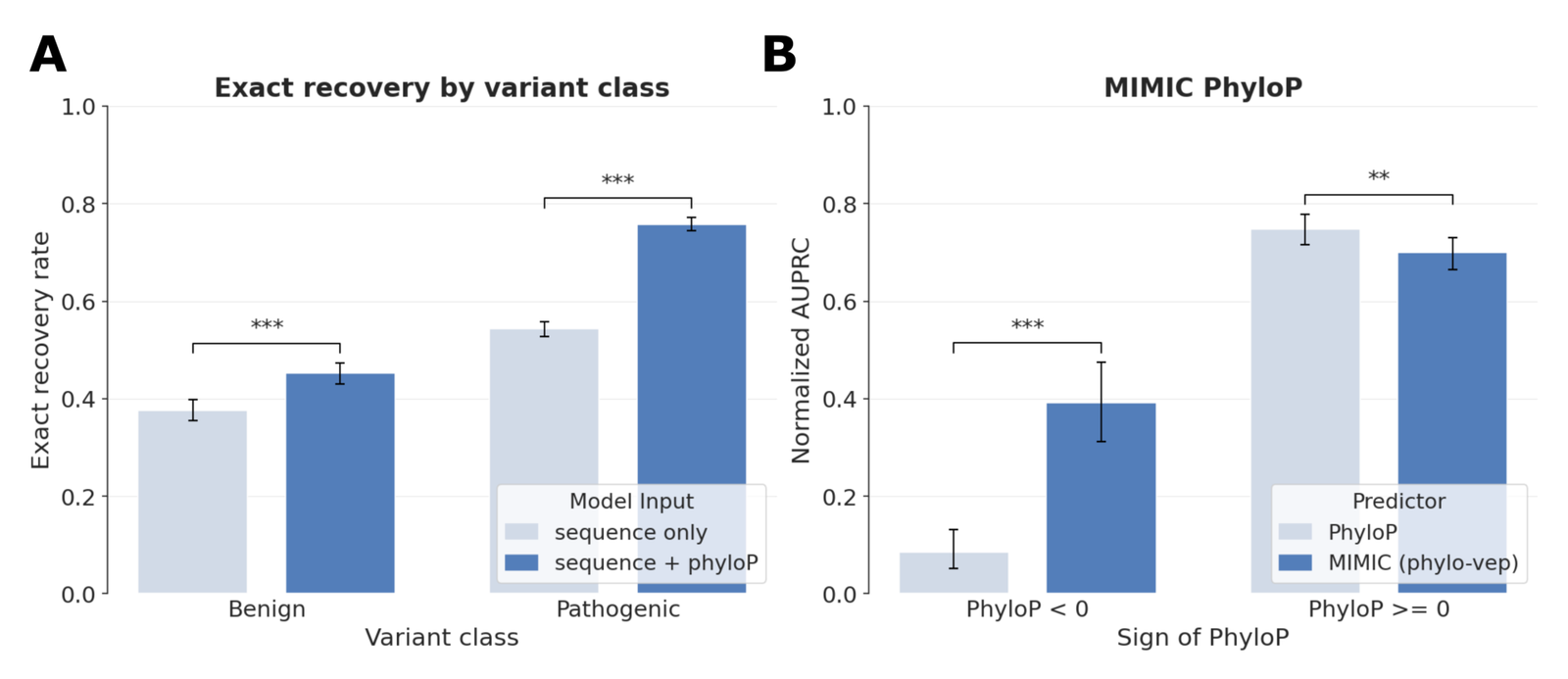 Figure 9: phyloP variant-effect prediction. (A) wild-type allele recovery from ClinVar pathogenic/benign SNVs, sequence-only vs sequence+phyloP. (B) normalized AUPRC split by sign of phyloP, raw phyloP vs MIMIC phylo-vep. Source: Golkar et al., MIMIC (arXiv:2604.24506, 2026), Supplementary Figure S5.
Figure 9: phyloP variant-effect prediction. (A) wild-type allele recovery from ClinVar pathogenic/benign SNVs, sequence-only vs sequence+phyloP. (B) normalized AUPRC split by sign of phyloP, raw phyloP vs MIMIC phylo-vep. Source: Golkar et al., MIMIC (arXiv:2604.24506, 2026), Supplementary Figure S5.
On complex variants the embedding-based VEP already leads (+33% vs SpliceBERT, the next-best), but the phyloP-based VEP vastly outperforms all embedding approaches (+82% over embedding-based MIMIC).106 On Mendelian variants, switching from embedding-based to phyloP-based scoring improves MIMIC by 90%.107 Underneath, MIMIC preferentially regenerates the wild-type allele when a pathogenic position is masked, and phyloP conditioning improves that recovery most for pathogenic variants, where evolutionary constraint is an especially strong signal.108
Limitations
LORE captures only a subset of the measurements that shape molecular phenotype; many important facets remain absent or weakly represented.109 The context scale is small relative to the biological processes one would want to capture, and the 1,000-token decoder window limits direct generation over long outputs.110 Splicing evaluation was restricted to genes under 25kb because many exceed the context length, so the strong AUPR numbers do not yet speak to long genes.76 The MaSIF surface modality covers only about 10% of structures, so surface-conditioned protein results lean on a thin slice of the data. Tellingly, the scarce-modality counts (phyloP 518,480, MaSIF 1,671,206, captions 176,626) are exactly the tracks the ablation found carry the most non-redundant signal.292426
The clinical and design results are entirely in silico: the HBB corrections validated against SpliceAI as an oracle, the binders against AlphaFold3 cofolding, not wet-lab assays. The honest negatives, the LBKWK half-life result where MIMIC trails by 8% and the hACE2 binder yield of 2 of 20, sit alongside the wins rather than being smoothed over.6994
There is also a reproducibility caveat. As of the preprint, MIMIC code and weights, plus LORE with its clustering, splits, and precomputed tokenization, were being prepared for release on the Polymathic AI GitHub under the MIT license, not yet fully downloadable.111112 The benchmark numbers above are the authors' own; the comparisons are set up carefully, with cluster-level splits and independent oracles, but independent replication will be the real test.
What it means
MIMIC's contribution is less any single benchmark and more a usable abstraction: treat sequence, structure, regulation, evolutionary constraint, and experimental context as different observations of one molecular state, then learn the joint distribution over it.113 The +1% to +3% gains over AlphaGenome and SpliceAI on coding-region splicing are modest; the value is that the same weights also predict splice sites, redesign an HBB transcript, fold an RNA, score a ClinVar variant, and produce a PD-L1 binder, collapsing five systems into one.778793
The broader claim is the one to watch. If heterogeneous aligned data is genuinely an alternative growth axis to raw scale, the next biological foundation models may compete on how many modalities they align rather than how many tokens they ingest. A 1B model beating a larger one on reconstruction is a single data point, not a law. For a practitioner with the artifacts in hand, the cheapest useful experiment is to feed MIMIC the extra aligned modalities you already have. Then check whether conditioning helps where your inputs are ambiguous from sequence alone. And when LORE ships, the most reusable artifact may not be the model at all. It may be the aligned dataset that made it possible.112
References
Footnotes
-
Golkar, S., Kovalic, J., Espejo Morales, I., et al. (2026). MIMIC: A Generative Multimodal Foundation Model for Biomolecules. arXiv:2604.24506. RNA 2D structure: sequence-alone F1 = 0.404 vs MIMIC-guided F1 = 0.987, nearly identical to the experimentally guided reference. https://arxiv.org/abs/2604.24506 ↩
-
Golkar et al. (2026), MIMIC. n = 1,000 RNA sequences; one-sided paired Wilcoxon signed-rank test, P = 6.25e-47. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC, abstract: LORE links nucleic acid, protein, evolutionary, structural, regulatory, and semantic/contextual modalities within partially observed biomolecular states. Submitted 27 April 2026, arXiv:2604.24506 [cs.AI]. https://arxiv.org/abs/2604.24506 ↩ ↩2
-
Polymathic AI (2026). MIMIC project entry and code/data release plan. https://github.com/PolymathicAI ; HuggingFace: https://huggingface.co/polymathicai/mimic ↩
-
Golkar et al. (2026), MIMIC, abstract: split-track encoder-decoder conditions on arbitrary subsets of observed modalities to reconstruct or generate missing components across genome, transcriptome, and proteome. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: at a comparable size to single-modality foundation models (1 billion parameters), MIMIC demonstrates the value of diverse and heterogeneous data as an alternative to pure scaling. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: the field has seen a proliferation of single-modality models that master one molecular modality but lack the capacity to operate across the broader central dogma. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: by focusing almost exclusively on forward prediction, these models fail to capture the full joint distribution of biological states. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: to understand a biological system, one must model the full joint probability distribution of its molecular states. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: given a desired protein structure, mRNA stability, and splicing pattern, what upstream nucleotide sequence is most likely to have produced them? arXiv:2604.24506. ↩
-
Abramson, J., Adler, J., Dunger, J., et al. (2024). Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630(8016):493-500. AlphaFold3 predicts the joint structure of complexes including proteins, nucleic acids, small molecules, ions, and modified residues within a single unified framework. https://doi.org/10.1038/s41586-024-07487-w ↩
-
Chen, J., Hu, Z., Sun, S., et al. (2022). Interpretable RNA Foundation Model from Unannotated Data for Highly Accurate RNA Structure and Function Predictions. arXiv:2204.00300. RNA-FM is a 12-layer transformer trained on 23 million ncRNA sequences via self-supervised learning. https://arxiv.org/abs/2204.00300 ↩
-
Golkar et al. (2026), MIMIC: approximately 15.5 million proteins and 13 million RNA transcripts from over 6,000 organisms, plus over 4 billion tokens of biomedical, functional, and contextual text. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: collected organisms span all domains of life and contain over 25 million transcripts and 123 billion nucleotides. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: full human and mouse genomes with GENCODE annotations, plus over 6,000 additional organisms from NCBI RefSeq. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: the central design principle of LORE is alignment, distinct from sequence-only corpora such as OpenGenome2 or BFD. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: each LORE row is an observation of a transcript, a protein, or a pairing of the two, containing the available subset of modalities. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: UniProt-to-RefSeq/Ensembl mapping yields a core subset of approximately 2 million aligned transcript/protein pairs. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: LORE is intentionally constructed as a partially observed multimodal dataset; no example is required to contain all modalities. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: approximately 150 distinct modality presence signatures across the dataset. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC, Table S1: nucleic acid sequence 12,967,153 samples. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC, Table S1: CAGE promoter usage 15,073,298 samples; ATAC-seq chromatin accessibility 14,021,301 samples. arXiv:2604.24506. ↩ ↩2
-
Golkar et al. (2026), MIMIC, Table S1: RNA chemical probing (RASP2) 1,644,404 samples. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC, Table S1: evolutionary conservation (phyloP) 518,480 samples. arXiv:2604.24506. ↩ ↩2
-
Golkar et al. (2026), MIMIC, Table S1: amino acid sequence, DSSP secondary structure, backbone tertiary structure, and SASA each 15,607,838 samples. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC, Table S1: MaSIF chemical surface 1,671,206 samples; protein abundance 1,803,028; functional captions 176,626. arXiv:2604.24506. ↩ ↩2 ↩3 ↩4
-
Golkar et al. (2026), MIMIC, Table S1: biomedical corpus 3,797,568 samples; 4 billion tokens after tokenization. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: phyloP conservation from 100-way alignments for human and 30-way for mouse. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: MaSIF surface features computed on about 10% of structures, roughly 50,000 CPU hours for 1.6M datasets. arXiv:2604.24506. ↩ ↩2
-
Golkar et al. (2026), MIMIC: MMseqs2 clustering with -c 0.8 and --min-seq-id 0.3 or 0.7, one representative per cluster. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC, Table S6: protein 30% identity 42,943,944 clusters from 299,566,796 members; RNA 30% identity 10,861,904 clusters from 25,001,445 members. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: cluster-level train/validation/test splits ensure no validation sequence shares significant similarity with training; 30% clusters define splits, 70% clusters ensure diversity. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: approximately 200M AlphaFoldDB v4 structures retained at pLDDT > 70. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: ESM3 structure tokenizer (VQ-VAE) with 4,096 classes based on local backbone geometry. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: built upon the 4M/AION encoder-decoder backbone. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: the split-track architecture sums aligned modalities while concatenating distinct molecular entities. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: the nucleic acid track element-wise sums sequence, structural data, and conservation scores into a single representation. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: split-track summation keeps total sequence length tractable regardless of modality count while preserving positional alignment between co-localized signals. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: encoder processes a unified token sequence of a nucleic acid track, an amino acid track, semantic/contextual token groups, and learnable register tokens. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: register tokens serve as information sinks pooling features across DNA, protein, and context tracks into a compact vector summary. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: all text-based modalities (context, functional captions, taxonomy) share a single embedding space with the biomedical corpus. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: RoPE with a local group-reset strategy; the position index restarts at zero for each track group. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: asymmetric context window, 10,000 tokens for the encoder and 1,000 for the decoder. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: decoupling observed context from predicted output allows conditioning on any observed subset and generating any target subset without architectural modification or task-specific output heads. arXiv:2604.24506. ↩
-
Jaegle, A., Borgeaud, S., Alayrac, J.-B., et al. (2022). Perceiver IO: A General Architecture for Structured Inputs & Outputs. ICLR 2022, arXiv:2107.14795. Flexible querying mechanism produces outputs of various sizes while scaling linearly with input and output size. https://arxiv.org/abs/2107.14795 ↩
-
Golkar et al. (2026), MIMIC, Table S7: encoder depth 20, decoder depth 12, hidden width 1,536, 24 attention heads per stack, 5 register tokens, RoPE fraction 0.75. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC, Table S7: self-attention masks are 50% bidirectional, 25% causal, 25% anti-causal, following the finding that causal attention can outperform bidirectional on certain tasks. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: training distributed by default across 92 NVIDIA H200 GPUs using NCCL. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC, Table S8: AdamW, betas (0.9, 0.95), weight decay 0.05, cosine learning-rate decay, bfloat16 precision. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: only the 566M-parameter encoder is used to generate embeddings; one of the smaller networks evaluated. arXiv:2604.24506. ↩ ↩2
-
Golkar et al. (2026), MIMIC: training defined as a stochastic sampling process over approximately 25 heuristic pathways, each specifying required and optional input and target modalities. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: staged curriculum scaling context from 1k to 2k to 4k to 8k to 10k tokens, learning local features before long-range dependencies. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: RNA/DNA are randomly cropped to fit the window because RNA folding is largely driven by local base-pairing. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: a protein whose structure track exceeds the budget is dropped rather than cropped, since a truncated protein cannot reliably predict structure. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: target-packing strategy fills the 1,000-token decoder budget for short or sparse targets. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: target packing acts as a regularizer, forcing the model to learn joint dependencies between splicing and conservation. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: register tokens trained via reconstruction with 0 to 10% random token dropout across all input tracks, preventing them from degenerating into copy mechanisms. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: dynamic workload balancing across GPUs to handle heavy-tailed sequence-length distributions, routing short sequences into large batches and long sequences into small ones (training systems, Section C.4). arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: a 100-token center window is masked and reconstructed in a single pass by sampling from a softmax at temperature 1e-8. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: random per-token reconstruction is about 0.25 for nucleotides and about 0.04 for amino acids. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: against ProtBERT, ESM-2, ESM-C, and ESM3-open, MIMIC conditioned with structural descriptors achieves the highest reconstruction accuracy. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: against NTv3 (masked) and Evo 2 (autoregressive) trained on OpenGenome2, MIMIC achieves the highest reconstruction accuracy in both intronic and exonic regions. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: even sequence-only, MIMIC is competitive with top sequence-only models, and conditioning on more modalities yields consistent improvements. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: on PFMBench (protein function, structure, interaction, developability; 11 tasks), MIMIC matches or exceeds ESM3, ESM-C, ProTrek, and SaProt, winning at least 7 of 11 tasks against each. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: strongest on protein-ligand binding (BindingDB, PDBbind) and best on all developability tasks, attributed to jointly pretraining on surface chemistry. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: on mRNABench, MIMIC outperforms Evo 2 and Orthrus on 4 of 7 tasks, a Dilated ResNet on 6 of 7, and all other methods on the remaining tasks. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: largest mRNABench gains on GO function (+2% vs Evo 2, +9% vs Orthrus) and protein localization (+2% vs Evo 2, +5% vs Orthrus). arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: Sugimoto half-life set, +8% relative improvement vs Orthrus. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: LBKWK half-life set, MIMIC trails by 8% vs Orthrus; benchmark from synthetic mRNA constructs where stability effects likely dominate. arXiv:2604.24506. ↩ ↩2
-
Golkar et al. (2026), MIMIC, Figure S3a: most effective single AA modalities are MaSIF hydrophobicity and tokenized backbone structure; combining all structural and chemical modalities beats any single one. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC, Figure S3b: largest intronic gains come from phyloP, splicing, and epigenetics (ATAC-seq or CAGE peaks). arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC, Figure S4: multimodal conditioning is most useful where the auxiliary modality is ambiguous from sequence alone rather than easily predictable; uplift peaks for informative-but-not-trivial modalities. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: about 99.85% of nucleotide positions are not splice sites, motivating AUPR. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: SpliceAI receives 5kb flanking on each side, AlphaGenome a 16,384-bp interval, MIMIC the full sequence up to 10kb. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: splice junction head outputs a 5-class distribution over non-splice, donor, acceptor, TSS, TES at each position. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: evaluation restricted to genes shorter than 25kb; genes over 10kb scored on a common 10kb region with the most splice junctions, because many genes exceed MIMIC's context length. arXiv:2604.24506. ↩ ↩2
-
Golkar et al. (2026), MIMIC, Figure 3a: gene-level splice AUPR +1% vs AlphaGenome, +3% vs SpliceAI, +11% vs NTv3 (coding); +12% vs AlphaGenome, +14% vs SpliceAI, +311% vs NTv3 (non-coding). arXiv:2604.24506. ↩ ↩2 ↩3
-
Golkar et al. (2026), MIMIC, Figure 3b: transcript-level splice AUPR +6% vs AlphaGenome, +5% vs SpliceAI, +11% vs NTv3 (coding); +26% vs AlphaGenome, +16% vs SpliceAI, +146% vs NTv3 (non-coding). arXiv:2604.24506. ↩ ↩2
-
Golkar et al. (2026), MIMIC: providing TSS and TES as input improves internal splice-structure recovery (+3% coding, +5% non-coding). arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: SPRY1, before conditioning MIMIC predicts an extra exon and alternative donor sites; after conditioning, false-positive junctions drop and only true exons remain. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: HBB c.316-197C>T (IVS-II-654 C>T), a canonical splice-altering mutation promoting pseudoexon inclusion through aberrant splice-site gain. arXiv:2604.24506. ↩
-
Nishikura, K. (2016). A-to-I editing of coding and non-coding RNAs by ADARs. Nature Reviews Molecular Cell Biology 17(2):83-96. A-to-I editing by ADAR enzymes can alter splice sites and recode coding sequence. https://doi.org/10.1038/nrm.2015.4 ↩
-
Golkar et al. (2026), MIMIC: phyloP VEP score distinguishes the pathogenic C>T from a matched non-splice-altering C>A control, localizing signal to the cryptic acceptor and donor. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: prompted with both the wild-type splice pattern and the pathogenic sequence with C>T fixed, generating designs for 30- or 50-nucleotide editable windows at varying distances. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: designs evaluated with SpliceAI as an independent oracle to avoid circularity with MIMIC-predicted splicing. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: produces designs that substantially reduce the likelihood of pathogenic splicing while maintaining the causal mutation. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: corrections found even when the editable window does not overlap the cryptic splice sites, demonstrating indirect regulatory rewiring through exonic splicing regulatory elements. arXiv:2604.24506. ↩ ↩2
-
Golkar et al. (2026), MIMIC: design targets PD-L1 (PDB 4ZQK chain A) and hACE2 (PDB 6VW1 chain A), the latter against the SARS-CoV-2 spike RBD. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: five conditioning strategies, 20 designs per strategy (backbone only; MaSIF surface for 40% or 100% of binding-site residues; backbone plus partial or full surface). arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: iterative design (up to 10 cycles, MIMIC for generation and verification); 200 hyperparameter sets per case filtered by ESMFold to top 20, then AlphaFold2 predicts folds, filtering to average pLDDT > 85 and scoring TM-score against native. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: design confidence peaks when both backbone structure and surface chemistry are provided; surface alone reaches pLDDT >= 85 for PD-L1, while the larger hACE2 relies more heavily on backbone conditioning. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: designed sequences recover the wild-type fold (median TM-score > 0.85) and surface chemistry (median MaSIF similarity > 0.90). arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: in silico co-folding with AlphaFold3, PD-L1, 35 of 37 high-confidence designs reach iPTM > 0.75 (median iPTM = 0.81). arXiv:2604.24506. ↩ ↩2
-
Golkar et al. (2026), MIMIC: AlphaFold3 cofolding, hACE2, only 2 of 20 high-confidence-fold designs predicted to bind the SARS-CoV-2 spike RBD at iPTM > 0.75. arXiv:2604.24506. ↩ ↩2
-
Golkar et al. (2026), MIMIC: generated designs maintain about 50% sequence identity to wild type. arXiv:2604.24506. ↩
-
Abramson et al. (2024), AlphaFold3: generative models are prone to hallucination, inventing plausible-looking structure even in unstructured regions. Nature 630. ↩
-
Golkar et al. (2026), MIMIC: instead of fixed output heads, MIMIC processes natural language descriptions of experimental conditions directly alongside molecular data, using context as semantic conditioning for assay-dependent chemical probing. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: correct experimental context significantly improves transcriptome-wide RASP2 predictions over sequence alone. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: MIMIC differentiates the broad sensitivity of in vivo icSHAPE (NAI-N3) from the A/C-restricted sensitivity of DMS-seq, generating condition-specific reactivity tracks. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: feeding MIMIC-predicted reactivity into ViennaRNA yields F1 = 0.987 vs F1 = 0.404 sequence-alone on the illustrated case, nearly identical to the experimentally guided reference. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: 1,000 held-out transcripts, three RNAfold (v2.7.2) predictions per transcript; median delta F1 = +0.061, mean +0.095 +/- 0.194 s.d. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: structural improvement significant at P = 6.25e-47 by a one-sided paired Wilcoxon signed-rank test. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: conditioning on phyloP improves RASP2 prediction, a structure-conservation synergy analogous to MSA coevolution in protein structure prediction. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: MIMIC exposes task-relevant signal directly through generation rather than forcing every task through a generic embedding. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: the phyloP-based VEP scores a variant by mean absolute perturbation in a 30-nucleotide window; the low-dimensional regressor is more interpretable and performs substantially better in practice. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: on complex variants the embedding-based VEP leads (+33% vs SpliceBERT), but the phyloP-based VEP outperforms it (+82% over embedding-based MIMIC). arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: on Mendelian variants, switching from embedding- to phyloP-based VEP improves performance by 90%. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: phyloP conditioning significantly increases correct wild-type allele recovery from ClinVar SNVs, more pronounced for pathogenic variants. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: LORE captures only a subset of measurements shaping molecular phenotype; many facets remain absent or weakly represented. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: current context scale remains limited, and the restricted 1,000-token decoder window limits direct generation over long outputs. arXiv:2604.24506. ↩
-
Golkar et al. (2026), MIMIC: code and weights, and LORE with clustering, splits, and precomputed tokenization, are being prepared for public release under the MIT license. arXiv:2604.24506. ↩
-
Polymathic AI (2026). MIMIC and LORE release on the Polymathic AI GitHub (MIT license). https://github.com/PolymathicAI/MIMIC ; HuggingFace: https://huggingface.co/polymathicai/mimic ↩ ↩2
-
Golkar et al. (2026), MIMIC: jointly modeling the interconnected layers of molecular biology through flexible multimodal representations of aligned molecular state. arXiv:2604.24506. ↩
Comments and feedback
Spotted an error or have a counterpoint? Comment below. No account needed, a name is enough. Corrections and pushback are welcome.