The most capable biomolecular structure predictor available, AlphaFold3, ships its model weights under a custom non-commercial license, and you cannot even download them. Academic and non-commercial researchers must request the parameters through a Google form, granted at DeepMind's sole discretion, with no redistribution permitted and outputs barred from training competing models.1 The model reported at least a 50% improvement over prior methods for predicting how proteins interact with other molecule types, with accuracy roughly doubled for some interaction categories,1 and most teams still cannot put it into anything they ship.
The reversal is sharper when you look at where the field came from. In January 2022, DeepMind relicensed the AlphaFold2 weights from non-commercial to CC BY 4.0, opening them to commercial use with attribution.2 Two years later, when AlphaFold3 shipped, the weights went the other way: research-only, gated, no redistribution.1 One model relicensed up, into the open. Its successor relicensed down, behind a gate.
So the question that actually matters when you sit down to fold something is not "what is the best model." It is "what am I folding, and what can I ship." Those two questions have different answers, and the gap between them is where most teams waste a quarter.
This is a survey of fifteen protein structure prediction foundation models, organized the way you would actually pick one: by task first, then by license. The set is AlphaFold2, AlphaFold-Multimer, AlphaFold3, Boltz-1, Boltz-2, Chai-1, ESMFold, HelixFold3, OmegaFold, OpenFold, Protenix, RoseTTAFold, RoseTTAFold All-Atom, RoseTTAFold2NA, and trRosetta. Where a lineage has a later successor that is not yet a standalone, evaluated release, such as OpenFold3 under the OpenFold project, it is mentioned as context rather than counted. Underneath the choices sit two real stories: an architectural one, the move from the Evoformer-era alignment models of 2020 through 2022 to the diffusion-era all-atom models of 2024 onward, and a licensing one, the split between AlphaFold2's open weights and AlphaFold3's gated ones. Both matter, but they are supporting context. The decision is the spine.
The master comparison
Here is every model in scope, with the facts that drive a decision. Bold names are introduced once here; the rest of the article refers back to this table.
| Model | Released | Params | Modality | License (weights) | Commercial? |
|---|---|---|---|---|---|
| AlphaFold2 | 20212 | ~93M2 | Single chain (MSA) | CC BY 4.02 | Yes |
| AlphaFold-Multimer | 2021.103 | undisclosed3 | Protein-protein | CC BY 4.03 | Yes |
| AlphaFold3 | 2024.051 | undisclosed1 | All-atom | Custom non-commercial1 | No |
| ESMFold | 20224 | ~15B total4 | Single chain (no MSA) | MIT4 | Yes |
| OmegaFold | 2022.075 | ~670M (PLM)5 | Single chain (no MSA) | Apache-2.05 | Yes |
| OpenFold | 20226 | ~93M6 | Single chain (MSA) | Apache-2.06 | Yes |
| Boltz-1 | 2024.117 | undisclosed7 | All-atom | MIT7 | Yes |
| Boltz-2 | 2025.068 | undisclosed8 | All-atom + affinity | MIT8 | Yes |
| Chai-1 | 2024.099 | undisclosed9 | All-atom | Apache-2.09 | Yes |
| Protenix | 2025.0110 | 368M (v1)10 | All-atom | Apache-2.010 | Yes |
| HelixFold3 | 2024.0811 | undisclosed11 | All-atom | Custom non-commercial11 | No |
| RoseTTAFold | 2021.0712 | undisclosed12 | Single chain + PPI | v1 non-commercial12 | v1 no |
| RoseTTAFold All-Atom | 2023.1013 | undisclosed13 | All-atom | BSD (caveats)13 | Mostly |
| RoseTTAFold2NA | 2022.0914 | ~67M14 | Protein-nucleic acid | MIT14 | Yes |
| trRosetta / trRosetta2 | 202015 | undisclosed15 | Single chain (MSA) | MIT code15 | Code yes |
Two columns deserve your attention before any benchmark does: license and modality. A model that wins every benchmark and that you cannot deploy is, for a commercial team, not a candidate. Keep that filter active while reading.
The decision, by task
Start with what is on your screen. The honest decision tree has six branches, and the license warning lives at the branch tips, not as a footnote.
 Decision logic synthesized from the per-model facts; license and capability claims for each leaf are cited in the corresponding section below.1243891114
Decision logic synthesized from the per-model facts; license and capability claims for each leaf are cited in the corresponding section below.1243891114
Single chain, deep MSA available
This is the classic case and the one with the clearest answer. If your target has many homologs and you can afford to build a multiple sequence alignment, AlphaFold2 remains the workhorse. On CASP14 it reached a median backbone accuracy of about 0.96 angstrom RMSD on the C-alpha atoms and a median domain GDT_TS of 92.4 out of 100, far ahead of every other group and approaching experimental accuracy.2 Its Evoformer trunk is a stack of 48 blocks using attention over an MSA representation and a residue-residue pair representation.2 The weights moved from non-commercial to CC BY 4.0 in January 2022, so commercial use is allowed with attribution.2
If you need to retrain, fine-tune, or audit the pipeline rather than just run it, reach for OpenFold instead. It is a trainable, memory-efficient PyTorch reimplementation of AlphaFold2 that matches AF2 accuracy on CAMEO and CASP-style validation when trained from scratch.6 It is Apache-2.0 for both code and weights, with no non-commercial restriction.6 OpenFold's training set, OpenProteinSet, ships about 400,000 MSAs and template hit files on the AWS open-data registry, which is why it is the model people actually fork.6 Its successor, OpenFold3, extends the same open posture to AF3-style cofolding, but as a project still maturing toward a fully evaluated release it sits outside the fifteen here.
trRosetta is the historical root of this branch and the honest "do not use this today" entry. It predicts inter-residue distances and orientation angles with a dilated ResNet of about 61 residual blocks, then folds with Rosetta energy minimization.15 It beat the AF1-era contact methods in 2019 and 2020,15 but it is two-stage rather than end-to-end and is superseded by RoseTTAFold and AlphaFold2.15 Worth knowing the lineage; not worth running.
Single chain, no MSA: orphans, antibodies, proteome scale
When alignment building is the bottleneck, or when your target has few homologs, the MSA itself becomes the problem rather than the solution. To see why that matters, look at what the alignment costs. A standard pipeline builds its MSA from UniRef30 at about 46 GB, BFD at about 272 GB, plus PDB100 structure templates at more than 100 GB, then runs a search over all of it before the network ever sees the sequence.12 RoseTTAFold folds a single structure in roughly ten minutes on one GPU once that alignment exists.12 The alignment is the slow part. Two models drop it entirely.
ESMFold is the one most people reach for. It replaces the MSA input with embeddings from a pretrained ESM-2 protein language model of up to 48 layers and 15 billion parameters.4 The folding head itself is only about 90 to 94 million parameters; the bulk is the language model.4 It folds a 384-residue protein in about 14.2 seconds on a single V100, up to 60 times faster than MSA-based predictors,4 and the team used it to predict more than 617 million metagenomic structures.4 The catch is accuracy: CAMEO average TM-score is about 0.83 and CASP14 about 0.68, below the full AlphaFold2 pipeline's roughly 0.88 and 0.85.4 It is MIT-licensed, including the weights.4
OmegaFold targets the same niche with a roughly 670-million-parameter language model called OmegaPLM feeding a geometry-aware transformer.5 On antibodies it reported RMSD of 2.12 angstrom versus AlphaFold2's 2.98 (p=0.0017), and on orphan proteins a TM-score of 0.73 versus 0.60 (p=0.0238).5 It is Apache-2.0,5 and runs about 10x faster than MSA-based pipelines.5 The caveat is maintenance: the repository has been dormant since 2022 and the niche has largely shifted to ESMFold.5 Treat it as a fast MSA-free baseline, not an actively developed tool.
The numbers make the trade concrete.
| Model | CAMEO TM-score | CASP14 TM-score | Speed vs MSA pipelines | Orphan / antibody evidence |
|---|---|---|---|---|
| AlphaFold2 (full MSA) | ~0.884 | ~0.854 | baseline (alignment-heavy)2 | weaker on orphans (shallow MSA)2 |
| ESMFold (single-seq) | ~0.834 | ~0.684 | up to 60x faster4 | folds orphans by design4 |
| OmegaFold (single-seq) | not reported5 | not reported5 | ~10x faster5 | orphan TM 0.73 vs 0.60; antibody RMSD 2.12 vs 2.985 |
Benchmark figures as reported by each model's primary publication; CAMEO and CASP14 are standard blind-assessment sets, and cross-paper comparisons should be read with care because evaluation sets and dates differ.
The practical read on this branch: single-sequence models do not beat a deep MSA on a well-studied family. They win when the alignment is shallow or absent, when you fold a whole proteome, or when latency is the constraint. That is a narrower claim than the hype around them suggested, and it is the honest one. Use ESMFold for the permissive license and the speed; keep OmegaFold as a fallback if you specifically want its antibody and orphan numbers and accept an unmaintained repo.
Protein-protein complexes
For protein-only complexes with known stoichiometry, AlphaFold-Multimer is the open default. It is the AlphaFold2 Evoformer and structure module retrained for multimeric inputs.3 On a benchmark of 17 template-free heterodimers it reached at least medium accuracy (DockQ at or above 0.49) on 13 of 17 targets and high accuracy (DockQ at or above 0.8) on 7 of 17, versus 9 and 4 for the prior state of the art.3 Its weights are CC BY 4.0, so it is commercially usable.3 Be explicit about its failure modes: it requires known stoichiometry, struggles with antibody-antigen and weak transient interactions, and can produce confident but wrong interfaces.3
If you want stronger multimer numbers and can run a single-sequence mode, Chai-1 reports a DockQ acceptable rate of about 69.8%, exceeding AlphaFold-Multimer 2.3's roughly 67.7%, and does so even without MSAs.9 More on Chai-1 in the all-atom branch; it doubles as a strong complex predictor.
All-atom: protein plus nucleic acid plus ligand
This is the branch AlphaFold3 defined and the one where the open ecosystem caught up fastest. The task is a single model that folds proteins, DNA, RNA, small-molecule ligands, ions, and modifications together.
AlphaFold3 sets the accuracy bar. It replaced the Evoformer with a Pairformer trunk of 48 blocks feeding a diffusion module that predicts raw atom coordinates directly.1 Its protein-ligand accuracy exceeds classical docking on PoseBusters without input pose information, its protein-nucleic-acid accuracy beats nucleic-acid-specific predictors, and its antibody-antigen accuracy markedly exceeds AlphaFold-Multimer.1 The headline doubling of accuracy on some interaction categories is real.1 The license is the problem. Code is Apache-2.0; weights are a custom non-commercial, research-only Terms of Use, must be requested from Google, cannot be redistributed, and outputs cannot be used to train competing models.1 For a commercial team, AlphaFold3 is a reference, not a dependency.
That gap is exactly why open reimplementations exist, and they are good.
 Lineage and license coloring synthesized from the per-model architecture and license facts cited throughout this article.7129111213
Lineage and license coloring synthesized from the per-model architecture and license facts cited throughout this article.7129111213
Boltz-1 was the first fully open, commercially usable model to approach AlphaFold3-level accuracy.7 It uses the AlphaFold3 architecture, an MSA module and Pairformer trunk feeding a diffusion module, and trains on PDB structures released before 30 September 2021, the same cutoff as AlphaFold3.7 On a recent-PDB test set its top-1 results were a mean LDDT of about 0.716, DockQ above 0.23 at about 0.625, and ligand RMSD below 2 angstrom at about 0.545, on par with AlphaFold3 and Chai-1 within bootstrap confidence intervals.7 It is MIT-licensed for both code and weights.7 That single licensing fact is why Boltz spread so fast.
Boltz-2 extends the lineage and is the current open state of the art.8 Beyond structure, it adds a binding-affinity module with dual heads for binary binding likelihood and continuous affinity regression.8 It is the first AI model to approach free-energy perturbation accuracy while being at least 1000 times cheaper, with a Pearson R of about 0.66 on the FEP+ 4-target subset and an out-of-the-box win on the CASP16 affinity challenge across 140 protein-ligand pairs.8 On hit discovery it reports an enrichment factor of 18.4 at 0.5% on MF-PCBA.8 It is MIT-licensed for model, weights, and code.8 If your question is "will this molecule bind, and how tightly," this is the open answer.
Chai-1 is the third open AlphaFold3-class model, Apache-2.0 for code and weights since a November 2024 relicensing.9 On PoseBusters protein-ligand it reports about 77% success, edging AlphaFold3's roughly 76%,9 and a CASP15 monomer C-alpha LDDT of about 0.849, above ESM3-98B's roughly 0.801.9 Its trick for strong single-sequence prediction is an extra input track of embeddings from a roughly 3-billion-parameter protein language model.9 Note the licensing history: at its September 2024 launch the weights were non-commercial, relicensed to Apache-2.0 in late November 2024, so verify the LICENSE in the repo for the version you pull.9
Protenix, from ByteDance, is an Apache-2.0 reproduction of AlphaFold3 with a disclosed parameter count: 368 million for v1 base.10 It is described as the first fully open-source model to surpass AlphaFold3 across multiple benchmark sets, with that superiority self-reported by the developers.10 It uses the AF3 pipeline of MSA and template embeddings, a Pairformer trunk, and a diffusion module.10 Default checkpoints use the 2021-09-30 cutoff to match AF3's training window.10
HelixFold3, from Baidu, is the open reimplementation to watch the license on. The architecture is an AlphaFold3-style Pairformer-plus-diffusion stack in PaddlePaddle.11 On antibody-antigen it surpasses AF3 in DockQ and success rate, exceeding 80% success with five specified epitope residues, and on PoseBusters ligands it is comparable to AF3 with over 90% stereochemistry pass rates.11 But the license is non-commercial: the README cites CC BY-NC-SA while the actual LICENSE file is a custom non-commercial license, an unresolved discrepancy, with commercial use requiring a separate paid Baidu offering.11 Strong model, restricted license. The same shape as AlphaFold3.
The Baker lab's RoseTTAFold All-Atom belongs in this branch too. It extends the three-track RoseTTAFold design with an atom-bond graph representation that treats ligands, ions, and modifications as flexible atoms and bonds rather than residue templates.13 On the CAMEO blind protein-ligand docking benchmark it outperforms classical docking such as AutoDock Vina and preserves ligand chemistry.13 The license is a BSD variant on code and weights, but with a real caveat: the bundled PDB100 template database is CC BY-NC-SA 4.0 and a runtime dependency.13 So the model is commercially usable but the default pipeline pulls in a restricted dependency. For broad complex prediction it is generally regarded as exceeded by AlphaFold3 and the Boltz and Chai reimplementations.13
Protein-nucleic acid, lightweight
If your task is specifically protein-DNA, protein-RNA, or RNA-only complexes and you do not want to stand up an AlphaFold3-class model, RoseTTAFold2NA is the purpose-built, lightweight choice. It is about 67 million parameters,14 a three-track network trained on PDB structures published before 30 May 2020 across roughly 26,128 protein clusters, 1,632 RNA clusters, and 1,556 protein-nucleic-acid complex clusters.14 On a 14-case held-out test it correctly identified 13 of 14 complexes versus 1 of 14 for AlphaFold-plus-docking baselines.14 It is MIT-licensed.14 Be honest about its ceiling: only about 29 to 30% of its complex predictions reach LDDT above 0.8, so the confidence estimate is needed to filter usable models.14 Use it as a fast first pass, not a final answer.
Need binding affinity
If the deliverable is a binding number rather than a structure, the branch collapses to one open answer: Boltz-2, covered above, with its affinity module approaching FEP accuracy at more than 1000x lower cost and an MIT license.8 No other model in this survey jointly predicts structure and affinity under an open license. That is a genuinely narrow field, and worth saying plainly.
The licensing split, stated plainly
Strip away the architecture talk and one practical question remains: can you put this model into the thing you are building. The answer splits cleanly, and not along the lines a leaderboard would suggest. The table below is the center of this survey. Read it before the benchmark numbers, because for most teams it filters the candidate set first.
| Model | Code license | Weights license | Commercial use? | Notable trap |
|---|---|---|---|---|
| AlphaFold2 | Apache-2.0 | CC BY 4.02 | Yes, with attribution2 | Relicensed from CC BY-NC to CC BY 4.0 in Jan 20222 |
| AlphaFold-Multimer | Apache-2.0 | CC BY 4.03 | Yes3 | Needs known stoichiometry; no ligands or nucleic acids3 |
| AlphaFold3 | Apache-2.0 | Custom non-commercial1 | No1 | Outputs may not be used to train competing models; weights gated, no redistribution1 |
| ESMFold | MIT | MIT4 | Yes4 | Successor ESM3 line is under different, partly non-commercial terms; do not assume ESMFold's MIT license carries over |
| OpenFold | Apache-2.0 | Apache-2.06 | Yes6 | None at the weights level6 |
| Boltz-1 | MIT | MIT7 | Yes7 | None at the weights level; superseded by Boltz-28 |
| Boltz-2 | MIT | MIT8 | Yes8 | None at the weights level; param count undisclosed8 |
| Chai-1 | Apache-2.0 | Apache-2.09 | Yes (since Nov 2024)9 | Launched non-commercial Sep 2024, relicensed Apache-2.0 Nov 20249 |
| Protenix | Apache-2.0 | Apache-2.010 | Yes10 | Benchmark wins over AF3 are self-reported10 |
| HelixFold3 | Custom non-commercial11 | Custom non-commercial11 | No (free tier)11 | README says CC BY-NC-SA, LICENSE file is custom; treat as restrictive11 |
| RoseTTAFold (v1) | MIT (code) | Rosetta-DL, non-commercial12 | No (weights)12 | RF2 and RFAA are permissive; only v1 weights are restricted12 |
| RoseTTAFold2NA | MIT | MIT14 | Yes14 | None at the weights level; inference databases carry own terms14 |
| RoseTTAFold All-Atom | BSD-style | BSD-style13 | Mostly13 | Bundled PDB100 templates are CC BY-NC-SA; SignalP6 dependency is academic-only13 |
| OmegaFold | Apache-2.0 | Apache-2.05 | Yes5 | Repo dormant since 20225 |
| trRosetta / trRosetta2 | MIT | Open15 | Code yes; pipeline no15 | Rosetta/PyRosetta refinement needs a paid commercial license15 |
Openness and commercial-use matrix, sorted from clean to restricted. "Commercial use?" answers the narrow question of whether the model's own weights permit it; the "Notable trap" column flags the per-model gotcha that the headline license hides. Sources cited inline per row.
Several patterns in that table are worth stating out loud, because they trip up real procurement decisions.
First, the headline model is the restricted one. AlphaFold3 weights are not openly downloadable; non-commercial researchers must request them through a Google form, granted at DeepMind's sole discretion, while commercial structure prediction is handled internally by Isomorphic Labs.1 That is the single most consequential licensing fact in the field right now.
Second, "open source" can hide a non-commercial dependency. trRosetta's network and weights are MIT, but the full folding pipeline depends on Rosetta and PyRosetta, free for academia and a paid license for for-profit use, so a commercial deployment of the complete pipeline is gated despite the MIT code.15 RoseTTAFold All-Atom has BSD-licensed code and weights, but its default pipeline pulls in a CC BY-NC-SA template database.13 A permissive license on the model file is necessary but not sufficient.
Third, licenses change, sometimes in your favor. Chai-1 launched in September 2024 under a restrictive non-commercial license, then Chai Discovery relicensed both code and weights to Apache-2.0 around late November 2024, which permits broad commercial use.9 AlphaFold2 made the same kind of move earlier, from CC BY-NC to CC BY in January 2022.2 Always check the LICENSE file at the version you are actually running.
Fourth, the same lab can ship across the fault line. The Baker lab's original RoseTTAFold distributes restricted, non-commercial weights under the Rosetta-DL license, but RoseTTAFold2 is MIT, RoseTTAFold2NA is MIT,14 and RoseTTAFold All-Atom is BSD-style.13 Within one lineage, only the v1 weights are the trap.
The clean cases, then, are AlphaFold2 and its open descendants: AlphaFold2 and AlphaFold-Multimer under CC BY 4.0,23 ESMFold under MIT,4 OpenFold under Apache-2.0,6 Boltz-1 and Boltz-2 under MIT,78 Chai-1 and Protenix under Apache-2.0,910 and RoseTTAFold2NA under MIT.14 The restricted cases are AlphaFold3,1 HelixFold3,11 and the original RoseTTAFold v1.12 The traps live in the caveats above. Read the dependencies, not just the LICENSE file.
Failure modes you inherit no matter which row you pick
Before the benchmark numbers, the failures behind them. These are shared across the family, and a high benchmark score on a curated set tells you nothing about whether your target hits one of them. Read the table in the next section with these in mind.
A high confidence score is not a correctness guarantee. AlphaFold-Multimer can produce confident but wrong interfaces and false-positive contacts.3 The confidence head is evidence, not proof.
The diffusion models hallucinate geometry. Boltz-1 produces overlapping and stacked identical chains in large complexes, a failure class it explicitly notes is shared with AlphaFold3.7 The newer Boltz-1x variant's inference-time steering largely fixes it, but the failure mode is real.7
Accuracy tracks MSA depth in the alignment-based family. AlphaFold2 has limited accuracy for orphan sequences that lack deep alignments,2 so a glowing benchmark on well-aligned targets says little about your low-homology one. This is the recurring tax across the MSA-based models, and the reason the single-sequence branch exists at all.
Disordered regions stay hard. AlphaFold2 and AlphaFold3 predict mostly static single conformations, not ensembles or dynamics, and degrade on intrinsically disordered regions.12 If your biology lives in a disordered region, a low confidence score there is telling you something true.
None of this is an edge case. It is the working condition. Wet-lab validation, not the confidence head, remains the arbiter.
The benchmark picture
Numbers across these models come from different papers, test sets, and metrics, so cross-model comparison is approximate. Treat the figures below as directional, and weight self-reported comparisons accordingly. With that stated, here is what each model reports on its own headline benchmark.
| Model | Headline benchmark | Reported result | License class |
|---|---|---|---|
| AlphaFold2 | CASP14 median GDT_TS2 | 92.4 / 100; ~0.96 A C-alpha RMSD2 | Permissive |
| AlphaFold-Multimer | 17 heterodimers, DockQ >= 0.493 | 13/17 medium, 7/17 high3 | Permissive |
| AlphaFold3 | Protein-other interactions1 | ~50%+ improvement, ~2x on some1 | Restricted |
| ESMFold | CAMEO / CASP14 TM-score4 | ~0.83 / ~0.68; ~60x faster4 | Permissive |
| OmegaFold | Antibody RMSD vs AF25 | 2.12 A vs 2.98 A (p=0.0017)5 | Permissive |
| OpenFold | CAMEO/CASP vs AF26 | matches AF2 from scratch6 | Permissive |
| Boltz-1 | Recent-PDB top-1 mean LDDT7 | ~0.716, on par with AF37 | Permissive |
| Boltz-2 | FEP+ affinity Pearson R8 | ~0.66, ~1000x cheaper than FEP8 | Permissive |
| Chai-1 | PoseBusters success9 | ~77% vs AF3 ~76%9 | Permissive |
| Protenix | Multiple AF3 benchmarks10 | reported to surpass AF3 (self-reported)10 | Permissive |
| HelixFold3 | SAbDab antibody-antigen11 | surpasses AF3; >80% with epitope hints11 | Restricted |
| RoseTTAFold2NA | 14-case held-out14 | 13/14 vs 1/14 baseline14 | Permissive |
Two patterns stand out. First, the open AlphaFold3 reimplementations cluster around AlphaFold3 itself, with differences often inside reported confidence intervals.79 Second, "surpasses AF3" claims for Protenix and on specific tasks for HelixFold3 are self-reported by the developers and benchmarked on their own chosen sets; treat them as promising rather than settled.1011 The benchmark winner and the model you can deploy are frequently not the same row, which is the entire reason to read by task and license rather than by score.
What changed under the hood: Evoformer to diffusion
The architectural shift maps cleanly onto a timeline, which is worth seeing before drawing conclusions.
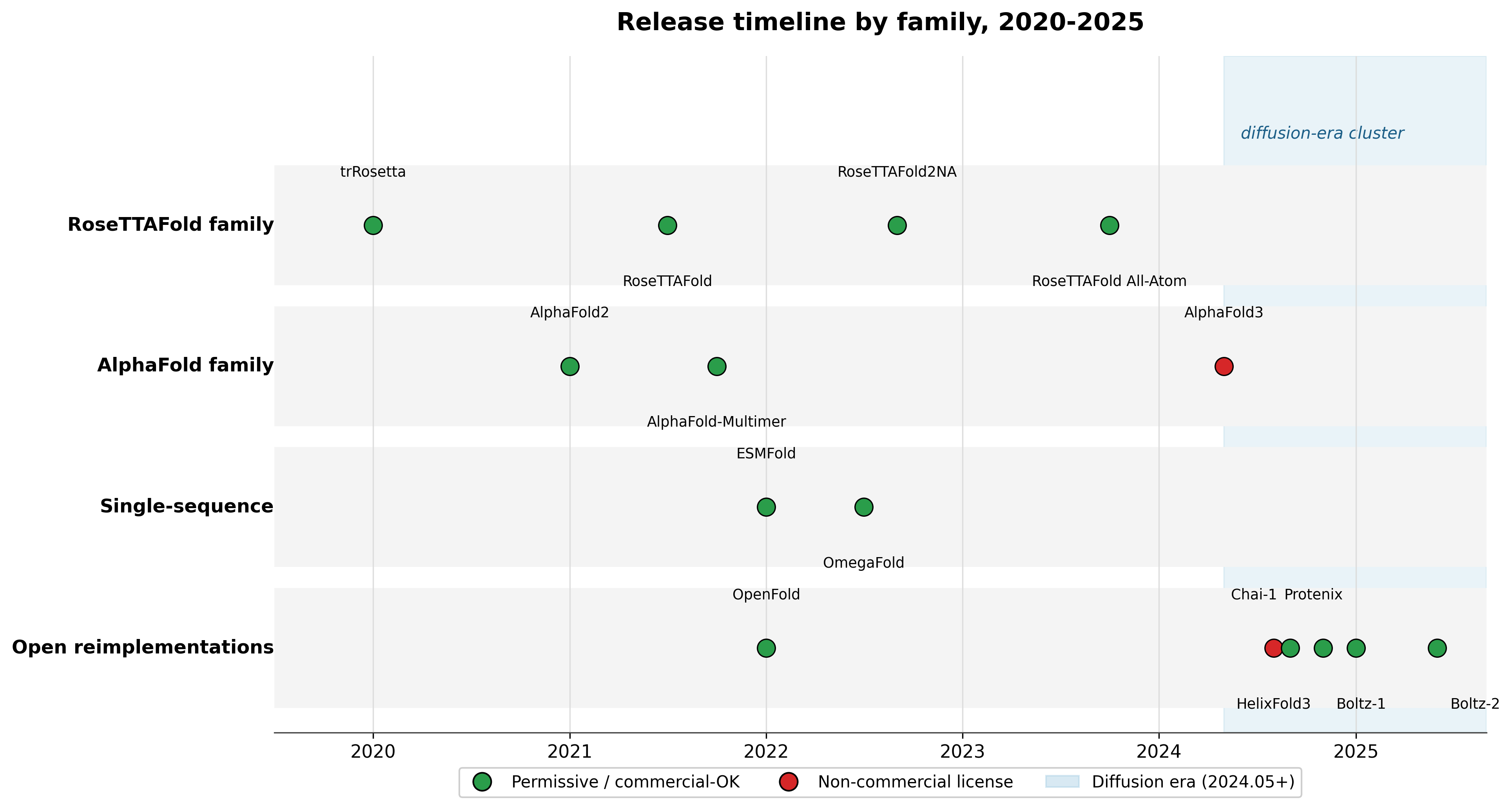 Release dates and license categories per the per-model facts; individual dates and licenses are cited in the sections above.712438911561012131415
Release dates and license categories per the per-model facts; individual dates and licenses are cited in the sections above.712438911561012131415
The first era, roughly 2020 to 2022, was built on multiple sequence alignments and the Evoformer. AlphaFold2's Evoformer was 48 blocks of attention over an MSA representation and a pair representation, feeding a structure module that produced coordinates as per-residue rotations and translations.2 RoseTTAFold reframed the same idea as a three-track network passing information between 1D sequence, 2D residue-pair, and 3D coordinate representations, approaching AF2-level accuracy on CASP14 while being faster.12 The whole era leaned on evolutionary information; accuracy tracked MSA depth, and orphan sequences suffered.2
The MSA-free models were the first crack in that assumption. ESMFold swapped the alignment for a protein language model's embeddings,4 and OmegaFold did the same with OmegaPLM.5 They traded accuracy for speed and independence from alignment building.
The second era, 2024 onward, replaced the Evoformer with a Pairformer and the structure module with diffusion. AlphaFold3's Pairformer has 48 blocks and reduced reliance on MSA processing, feeding a diffusion module that denoises raw atom coordinates directly.1 That architecture is what unlocked unified all-atom prediction across proteins, nucleic acids, ligands, and modifications in one model.1 Every open AlphaFold3-class model here, Boltz-1, Boltz-2, Chai-1, Protenix, and HelixFold3, copies that Pairformer-plus-diffusion shape.7891011 The architecture became a commodity within a year. What did not become a commodity was the original's weights.
What we do not know
Being explicit about the gaps is part of a fair survey.
Parameter counts are undisclosed for most of these models. AlphaFold3, AlphaFold-Multimer, Boltz-1, Boltz-2, Chai-1, HelixFold3, RoseTTAFold, and RoseTTAFold All-Atom all decline to state a total count.13789111213 We have firm numbers only for AlphaFold2 at about 93 million,2 RoseTTAFold2NA at about 67 million,14 Protenix-v1 at 368 million,10 and ESMFold's folding head at about 90 to 94 million on top of its language model.4 Scaling claims for the undisclosed models cannot be independently checked.
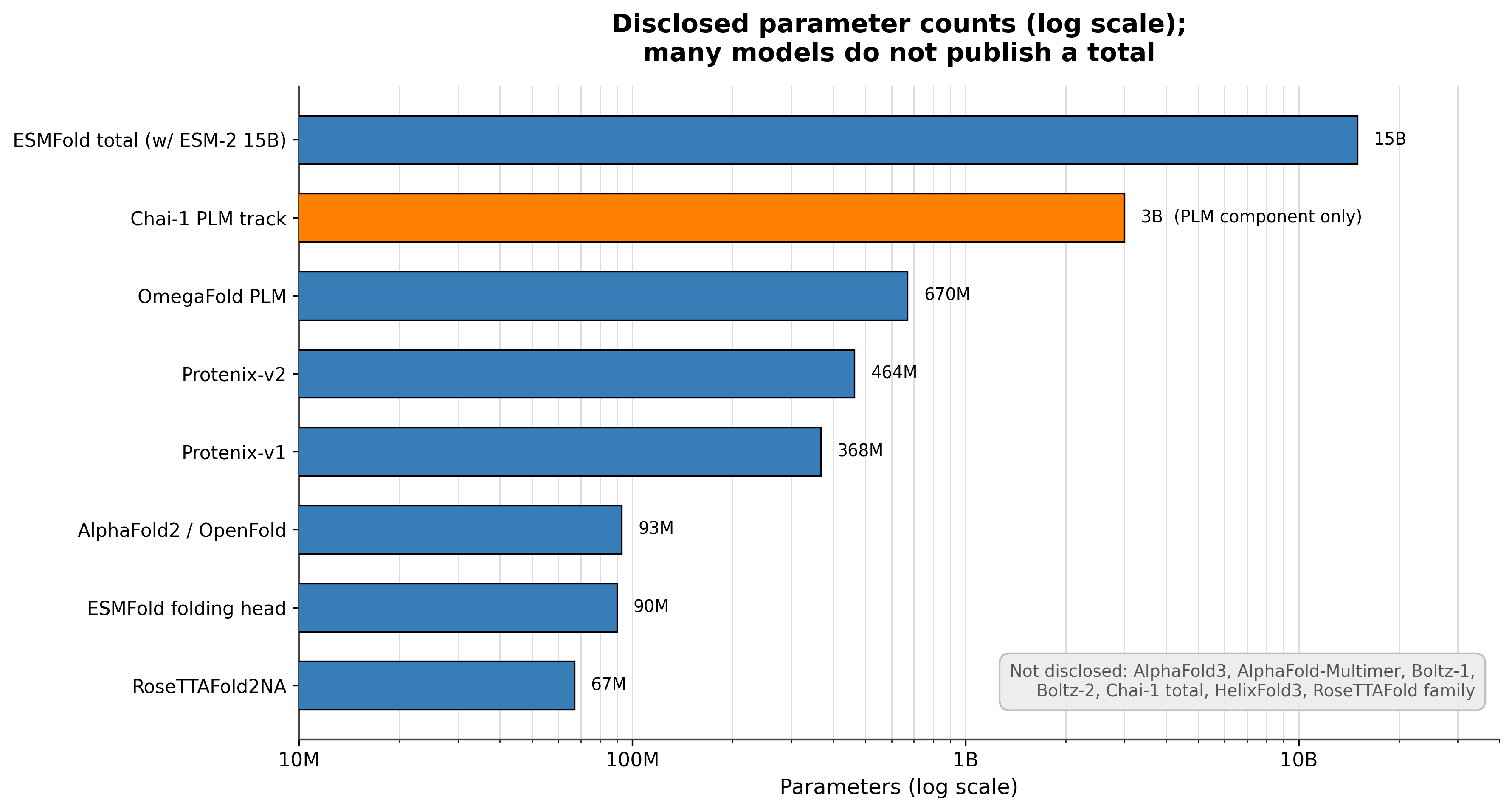 Disclosed parameter counts, log scale. Only published numbers are plotted; the list of undisclosed models is the more telling data point. Sources: AlphaFold2,2 RoseTTAFold2NA,14 OpenFold,6 ESMFold,4 Protenix,10 OmegaFold,5 Chai-1.9
Disclosed parameter counts, log scale. Only published numbers are plotted; the list of undisclosed models is the more telling data point. Sources: AlphaFold2,2 RoseTTAFold2NA,14 OpenFold,6 ESMFold,4 Protenix,10 OmegaFold,5 Chai-1.9
Cross-model benchmark comparison is shaky. These numbers come from different papers, test sets, cutoffs, and metrics. The "surpasses AlphaFold3" claims from Protenix and HelixFold3 are self-reported on developer-chosen benchmarks.1011 AlphaFold3 itself published a December 2024 addendum correcting some reported metrics.1 Independent head-to-head evaluation on a single shared test set is what would settle the ranking, and it does not yet exist for this full set.
The shared failure modes laid out earlier, the confident-but-wrong interfaces, the stacked-chain hallucinations, the MSA-depth dependence, and the weakness on disordered regions, are underexamined precisely because no shared benchmark stresses them in one place.3712 They are the working conditions, not edge cases, and wet-lab validation remains the arbiter.
How to actually choose
Strip it to the decisions that hold up.
If you have a single chain with deep homolog coverage and need maximum accuracy, run AlphaFold2; it is open and still the single-chain leader.2 If you need to fork, retrain, or audit, use OpenFold.6
If alignment building is your bottleneck, or you are folding orphans or working at proteome scale, use ESMFold for the permissive license and the speed, with OmegaFold as a fallback.45
If you need protein-protein complexes and want open weights, AlphaFold-Multimer is the default,3 with Chai-1 a stronger and also open alternative.9
If you need all-atom prediction across proteins, nucleic acids, and ligands and you can ship commercially, the answer is one of the open AlphaFold3 reimplementations: Boltz-2 if you also want affinity,8 Chai-1 or Protenix otherwise.910 AlphaFold3 and HelixFold3 are more capable on specific tasks but non-commercial.111
If you need protein-nucleic-acid structures and want something light, RoseTTAFold2NA, with its confidence filter on.14
If you need a binding number, Boltz-2.8
The model that wins the benchmark and the model you can deploy are usually different rows in the table. AlphaFold3 is the most capable predictor published, and for a commercial team it is mostly a reference point. The open reimplementations closed the accuracy gap inside a year; the license gap is the one that still decides projects. The frontier is gated. The usable frontier, increasingly, is not. Pick by what you are folding and what you can ship, and the fifteen models collapse into a short, defensible shortlist.
References
Footnotes
-
Abramson, Adler, Dunger et al., "Accurate structure prediction of biomolecular interactions with AlphaFold 3," Nature 630, 493-500 (2024). https://www.nature.com/articles/s41586-024-07487-w ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20 ↩21 ↩22 ↩23 ↩24 ↩25 ↩26 ↩27
-
Jumper et al., "Highly accurate protein structure prediction with AlphaFold," Nature 596:583-589 (2021). https://doi.org/10.1038/s41586-021-03819-2 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20 ↩21 ↩22 ↩23 ↩24 ↩25 ↩26 ↩27
-
Evans et al., "Protein complex prediction with AlphaFold-Multimer," bioRxiv 2021 (updated 2022). https://www.biorxiv.org/content/10.1101/2021.10.04.463034v2 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19
-
Lin et al., "Evolutionary-scale prediction of atomic-level protein structure with a language model," Science 379:6637 (2023), pp.1123-1130. https://www.science.org/doi/10.1126/science.ade2574 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20 ↩21 ↩22 ↩23 ↩24 ↩25 ↩26
-
Wu et al., "High-resolution de novo structure prediction from primary sequence," bioRxiv 2022. https://doi.org/10.1101/2022.07.21.500999 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20 ↩21
-
Ahdritz, G. et al., "OpenFold: retraining AlphaFold2 yields new insights into its learning mechanisms and capacity for generalization," Nature Methods 21, 1514-1524 (2024). https://www.nature.com/articles/s41592-024-02272-z ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15
-
Wohlwend, Corso, Passaro et al., "Boltz-1: Democratizing Biomolecular Interaction Modeling," bioRxiv 2024. https://www.biorxiv.org/content/10.1101/2024.11.19.624167 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20
-
Passaro, Corso, Wohlwend et al., "Boltz-2: Towards Accurate and Efficient Binding Affinity Prediction," bioRxiv 2025. https://www.biorxiv.org/content/10.1101/2025.06.14.659707v1 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20 ↩21 ↩22
-
Chai Discovery team, "Chai-1: Decoding the molecular interactions of life," bioRxiv 2024. https://doi.org/10.1101/2024.10.10.615955 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20 ↩21 ↩22 ↩23 ↩24 ↩25
-
ByteDance Protenix Team, "Protenix - Advancing Structure Prediction Through a Comprehensive AlphaFold3 Reproduction," bioRxiv 2025. https://www.biorxiv.org/content/10.1101/2025.01.08.631967v1 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20
-
PaddleHelix Team, Baidu, "Technical Report of HelixFold3 for Biomolecular Structure Prediction," arXiv:2408.16975 (2024). https://arxiv.org/abs/2408.16975 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20 ↩21
-
Baek M. et al., "Accurate prediction of protein structures and interactions using a three-track neural network," Science 373:871-876 (2021). https://www.science.org/doi/10.1126/science.abj8754 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13
-
Krishna R., Wang J., Ahern W. et al., "Generalized biomolecular modeling and design with RoseTTAFold All-Atom," Science 384, eadl2528 (2024). https://www.science.org/doi/10.1126/science.adl2528 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15
-
Baek, McHugh, Anishchenko, Jiang, Baker, DiMaio, "Accurate prediction of protein-nucleic acid complexes using RoseTTAFoldNA," Nature Methods (2023). https://www.nature.com/articles/s41592-023-02086-5 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20
-
Yang J, Anishchenko I, Park H, Peng Z, Ovchinnikov S, Baker D, "Improved protein structure prediction using predicted interresidue orientations," PNAS 117(3):1496-1503 (2020). https://doi.org/10.1073/pnas.1914677117 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11
Comments and feedback
Spotted an error or have a counterpoint? Comment below. No account needed, a name is enough. Corrections and pushback are welcome.