ESMFold predicts atomic-level structure from a single sequence. No multiple sequence alignment, no database search, no Evoformer churning over homologs. You hand it a string of amino acids and it returns coordinates and a per-residue confidence.1
That is the whole trick, and it should bother you. Every accurate structure predictor of the AlphaFold2 era leaned on evolutionary information packed into multiple sequence alignments.2 Coevolving residues tend to be in contact; the MSA carries the signal; the network reads it off. Remove the MSA and you remove the signal. So how does a model fold a sequence it has never seen, with no alignment to lean on?
The answer is that the alignment never left. It moved inside the weights of a language model. The MSA was the access mechanism, not the information. Scale a language model on raw protein sequence to 15 billion parameters and "an atomic-resolution picture of protein structure emerges in the learned representations."3 The alignment was a workaround for a model that was not yet big enough.
The MSA Is the Bottleneck
Anfinsen won a Nobel prize in 1972 for demonstrating a connection between a protein's amino acid sequence and its three-dimensional structure.4 Sequence determines structure. The hard part has always been computing the map. Deep learning methods took center stage at CASP14, with AlphaFold2 achieving remarkable accuracy,5 and RoseTTAFold followed with a three-track network processing sequence, distance, and coordinate information simultaneously, approaching those accuracies.6
Both lean on evolutionary information. AlphaFold2 and RoseTTAFold each build an MSA, extract the coevolutionary signal, and feed it to a structure module. The signal is real and powerful. Coevolving residue pairs tend to be in physical contact, which is precisely the constraint a structure module wants.
The cost is the alignment. You search large sequence databases per query, build the MSA, and only then run the network. For orphan sequences with few homologs, the MSA is shallow and the signal degrades. At metagenomic scale, where you have hundreds of millions of sequences and many lack close relatives, the search dominates wall-clock time. ESMFold removes that stage entirely. It "does not require any lookup or MSA step, and therefore does not require any external databases to be present in order to make predictions."1 The two pipelines diverge at step one.
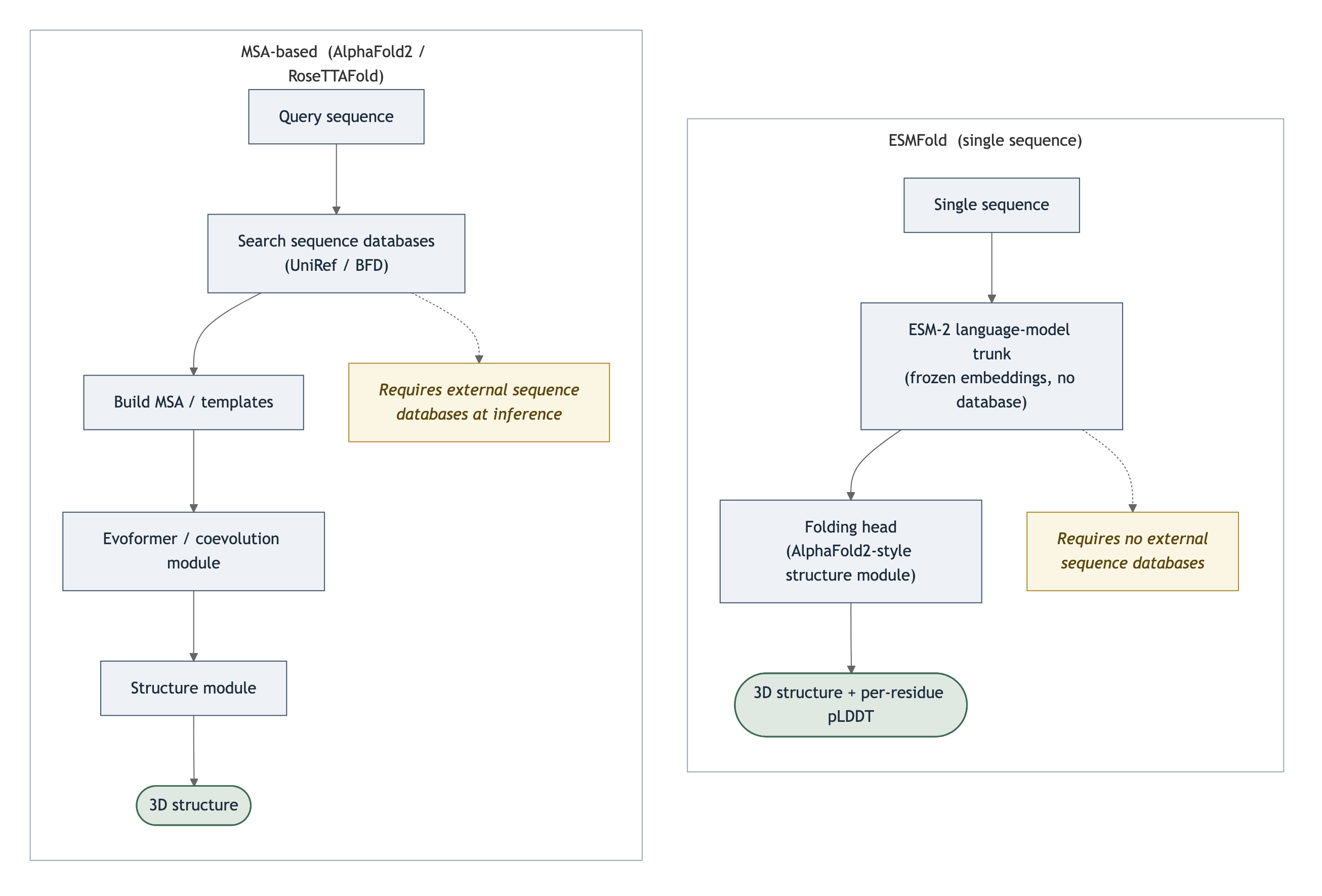 Figure 1: The MSA-based pipeline searches external databases and aligns homologs before folding. ESMFold replaces the whole alignment stage with the ESM-2 trunk and reads structure off single-sequence representations. Source: Lin et al., Science 2023, DOI 10.1126/science.ade2574; ESMFold model card, facebook/esmfold_v1.
Figure 1: The MSA-based pipeline searches external databases and aligns homologs before folding. ESMFold replaces the whole alignment stage with the ESM-2 trunk and reads structure off single-sequence representations. Source: Lin et al., Science 2023, DOI 10.1126/science.ade2574; ESMFold model card, facebook/esmfold_v1.
Structure Falls Out of Scale
ESM-2 is a transformer trained on a masked language modeling objective over protein sequences.7 Mask a residue, predict it from context, repeat across hundreds of millions of UniRef sequences. No structural labels, no contacts, no coordinates in the loss. So why should masked-token prediction encode geometry at all? Because the identity of a buried residue is constrained by the residues it touches in three dimensions, not just by its neighbors in sequence. A hydrophobic core, a salt bridge, a disulfide pair: each is a statistical regularity that shows up in which amino acids co-occur. To fill in a masked residue well, the model has to respect the constraints that make a sequence foldable in the first place. The cheapest way to lower masked-token loss is to learn the statistics that structure imposes, so the optimizer finds them.
This thesis predates ESM-2. The predecessor, ESM-1b (Rives et al., 2021), trained a contextual language model on 250 million sequences and found that "without prior knowledge, information emerges in the learned representations on fundamental properties of proteins such as secondary structure, contacts, and biological activity."8 ESM-1b is a single-sequence model, 650M parameters, and it already improved state-of-the-art features for long-range residue-residue contact prediction.9 The signal was there. It just was not strong enough to skip the alignment.
ESM-2 pushes the same objective much further, up to 15 billion parameters.3 Structure was never trained for. It precipitated out as a side effect of predicting masked residues well. The mechanism for reading it back out is simple enough to be suspicious. For unsupervised contact prediction, "a sparse linear combination of the attention heads is used to directly predict protein contacts, fitted with logistic regression on 20 structures."10 Twenty structures to calibrate a linear probe. The attention maps already encode the contacts; the probe just points at them. Long-range here means a sequence separation of at least 24 residues, scored as top-L,LR precision.11
The Family Scales Monotonically
ESM-2 ships as six checkpoints, and the spread is the argument. Same masked-LM objective, same data family (UR50/D 2021_04), four orders of magnitude apart in parameters.
| Checkpoint | Layers | Parameters | Embedding dim |
|---|---|---|---|
| esm2_t6_8M_UR50D | 6 | 8M | 320 |
| esm2_t12_35M_UR50D | 12 | 35M | 480 |
| esm2_t30_150M_UR50D | 30 | 150M | 640 |
| esm2_t33_650M_UR50D | 33 | 650M | 1280 |
| esm2_t36_3B_UR50D | 36 | 3B | 2560 |
| esm2_t48_15B_UR50D | 48 | 15B | 5120 |
Table 1: The ESM-2 checkpoint family. Larger sizes "generally have somewhat better accuracy, but require much more memory and time to train." Source: HuggingFace ESM-2 model cards; ESM GitHub README, facebookresearch/esm.
Two metrics track the size, and it is worth keeping them apart. The first is unsupervised long-range contact precision: the linear probe on attention heads, scored top-L,LR at >=24 residue separation. The second is end-to-end structure-prediction accuracy: the full folding model's agreement with experimental structures. They are different measurements on different scales.
Contact precision climbs monotonically with scale. On the CAMEO set it rises from 15.7 at 8M to 51.7 at 15B; on CASP14, from 9.8 to 37.0.12 The curve has not flattened: the 15B model still beats the 3B. And the single-sequence ESM-2 catches its own predecessor early. ESM-2 at 150M already reaches 42.2 long-range contact precision on the large-validation set, edging past ESM-1b at 650M (41.1).13 More parameters on a smaller single-sequence model beat fewer parameters on a larger one, same training signal.
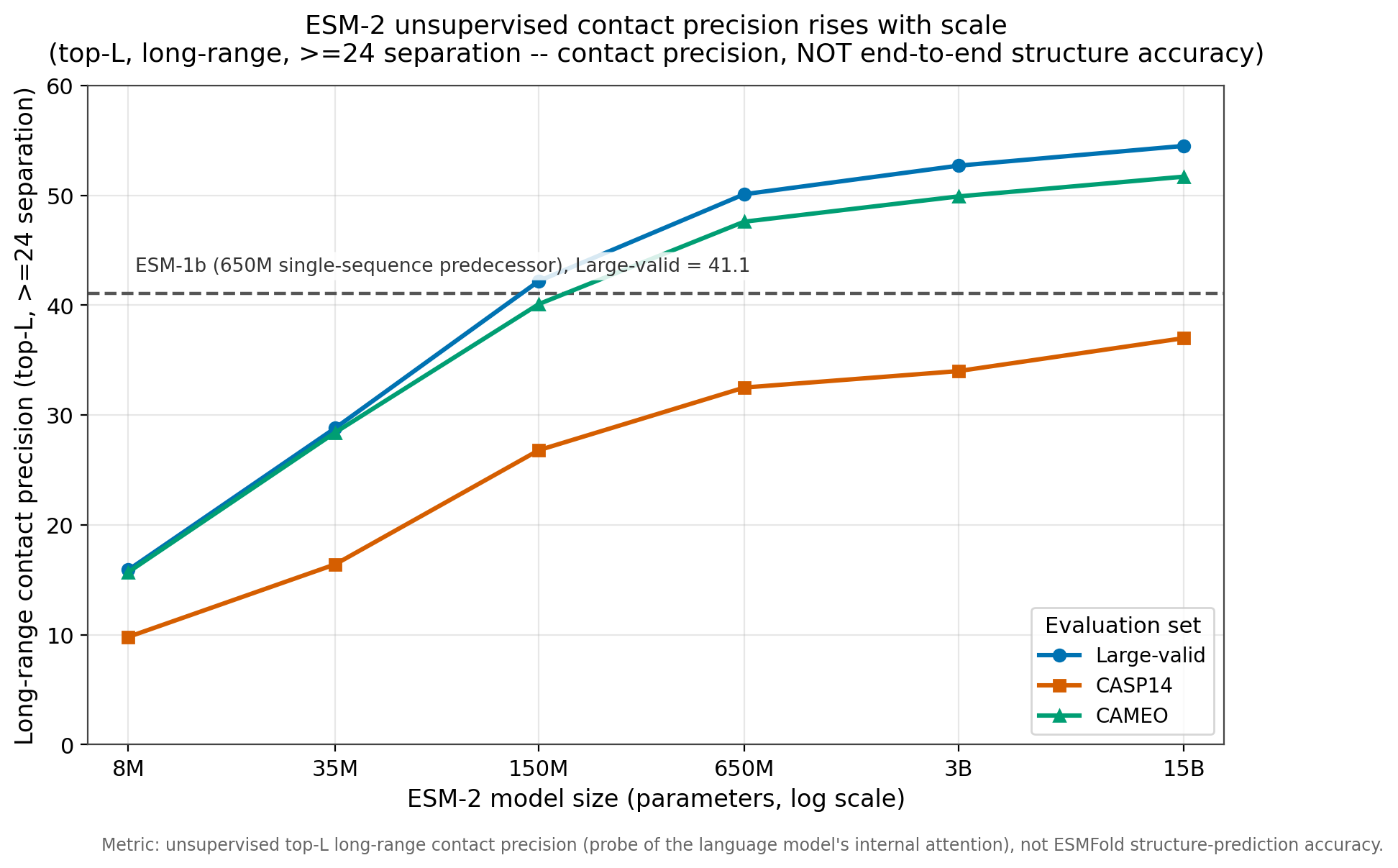 Figure 2: Unsupervised long-range contact precision (top-L,LR) for the ESM-2 family. This is the contact-precision metric, distinct from end-to-end structure accuracy. The 650M and 700M labels refer to the same checkpoint. Source: ESM GitHub README comparison table, facebookresearch/esm.
Figure 2: Unsupervised long-range contact precision (top-L,LR) for the ESM-2 family. This is the contact-precision metric, distinct from end-to-end structure accuracy. The 650M and 700M labels refer to the same checkpoint. Source: ESM GitHub README comparison table, facebookresearch/esm.
End-to-end structure prediction tracks the same scaling, on its own scale. On CASP14, accuracy rises from 36.7 at 8M to 55.4 at 15B; on CAMEO, from 48.1 to 72.1.14 One number anchors the emergence claim. The 15B model reaches 72.1 on CAMEO from single sequences alone. For contrast, the MSA Transformer (ESM-MSA-1b), which consumes alignments rather than single sequences, posts a large-validation contact precision of 57.4 against ESM-2 15B's 54.5 on the same column.15 The single-sequence model is no longer obviously paying a fatal penalty for dropping the MSA. It learned the alignment instead of reading it.
A Folding Head on a Frozen Trunk
ESMFold is "a state-of-the-art end-to-end protein folding model based on an ESM-2 backbone."16 The construction is deliberately thin: "for structure prediction, an AlphaFold2 structure module is trained directly from the frozen language model embeddings."17 The expensive part, the representation, is already done by ESM-2. The language model stays frozen; only the folding head learns to map embeddings to coordinates, end to end, directly from the sequence of a protein.18
The recommended checkpoint is esmfold_v1: a 690M-parameter folding head sitting on top of the 3B-parameter ESM-2 trunk, written in the README's table as 48 (+36) layers and 690M (+3B) parameters.19 The earlier esmfold_v0 was the checkpoint used for the experiments in the Lin et al. paper.20 By default the model runs 4 recycles, the number used in training,21 and on long sequences it "chunks axial attention computation to reduce memory usage from O(L^2) to O(L)."22 Confidence comes back per residue as pLDDT, stored in the B-factor field of the output, exactly as in AlphaFold2.23
The obvious objection is memorization: maybe the model just recalls folds it saw in training. Generalization is measured the hard way to rule that out. The structural split dataset implements holdouts at the family, superfamily, and fold level, using the SCOPe database to classify domains.24 A family-level holdout removes close relatives; a fold-level holdout removes everything sharing the same overall topology. The fold-level case is the strict one: the model must predict a structure whose fold it never observed during training, with no near-neighbor to interpolate from. ESMFold is evaluated against those holdouts, not just against close relatives of training examples, which is what makes the emergence claim about the representation rather than about the training set.
Dropping the MSA removes the slowest stage of the pipeline. The result, in the abstract's words, is "an order-of-magnitude acceleration of high-resolution structure prediction."25 The model card puts it qualitatively: inference time is "very significantly faster than AlphaFold2."26 That speed is not a convenience. It is the gate to the last section.
| ESMFold (single sequence) | AlphaFold2 (MSA-based) | |
|---|---|---|
| Input | One sequence1 | Sequence plus MSA2 |
| MSA / external DB at inference | None required1 | Required2 |
| Representation | Frozen ESM-2 embeddings17 | Evoformer over MSA |
| Relative inference speed | Order-of-magnitude faster25 | Baseline |
| Per-residue confidence | pLDDT23 | pLDDT |
Table 2: ESMFold versus the MSA-based pipeline. Only attributes supported by the verified sources are filled. Source: Lin et al., Science 2023, DOI 10.1126/science.ade2574; HuggingFace ESMFold model card; ESM GitHub README, facebookresearch/esm.
Running It
There are two paths to a prediction. If you do not want to host a 3-billion-parameter backbone, the ESM Metagenomic Atlas exposes a fold API; you POST a single sequence over HTTP and get back a PDB.27
curl -X POST \
--data "MKTAYIAKQRQISFVKSHFSRQLEERLGLIEVQAPILSRVGDGTQDNLSGAEKAVQVKVKALPDAQF" \
https://api.esmatlas.com/foldSequence/v1/pdb/
That one POST returns a folded structure. For local or batch work, load ESMFold through the HuggingFace transformers library;28 the model card calls this the easy entry point, and the folding model wants Python 3.9 or earlier with PyTorch installed.29 The per-residue confidence lands in the PDB B-factor column.23
from transformers import AutoTokenizer, EsmForProteinFolding
import torch
tokenizer = AutoTokenizer.from_pretrained("facebook/esmfold_v1")
model = EsmForProteinFolding.from_pretrained("facebook/esmfold_v1")
seq = "MKTAYIAKQRQISFVKSHFSRQLEERLGLIEVQAPILSRVGDGTQDNLSGAEKAVQVKVKALPDAQF"
inputs = tokenizer([seq], return_tensors="pt", add_special_tokens=False)
with torch.no_grad():
outputs = model(**inputs)
pdb = model.output_to_pdb(outputs)[0] # list[str]; pLDDT lands in the B-factor column
with open("prediction.pdb", "w") as handle:
handle.write(pdb)
No database path, no MSA object, no alignment step. The sequence goes in, the PDB comes out. For embeddings rather than folds, ESM-2 itself is "suitable for fine-tuning on a wide range of tasks that take protein sequences as input,"30 and the README notes it can serve variant-effect prediction with performance expected to be similar to the specialized ESM-1v.31 The 650M checkpoint (facebook/esm2_t33_650M_UR50D) is the practical default; the 15B model can hit out-of-memory errors on a single GPU, which the README addresses with Fairscale FSDP CPU offloading.32
The Payoff Is Metagenomic
Accuracy gets cited; speed is what changes the science. An order-of-magnitude faster predictor that needs no MSA can run on sequences that have no homologs to align against, which is exactly the situation across the metagenomic world. That acceleration "enables large-scale structural characterization of metagenomic proteins."25
So Meta folded the metagenome. The authors applied ESMFold to "construct the ESM Metagenomic Atlas by predicting structures for >617 million metagenomic protein sequences, including >225 million that are predicted with high confidence."33 The v0 release in November 2022 opened the full 617 million predicted structures.34 A March 2023 update with EBI, v2023_02, added another 150 million predicted structures plus pre-computed ESM-2 embeddings.35
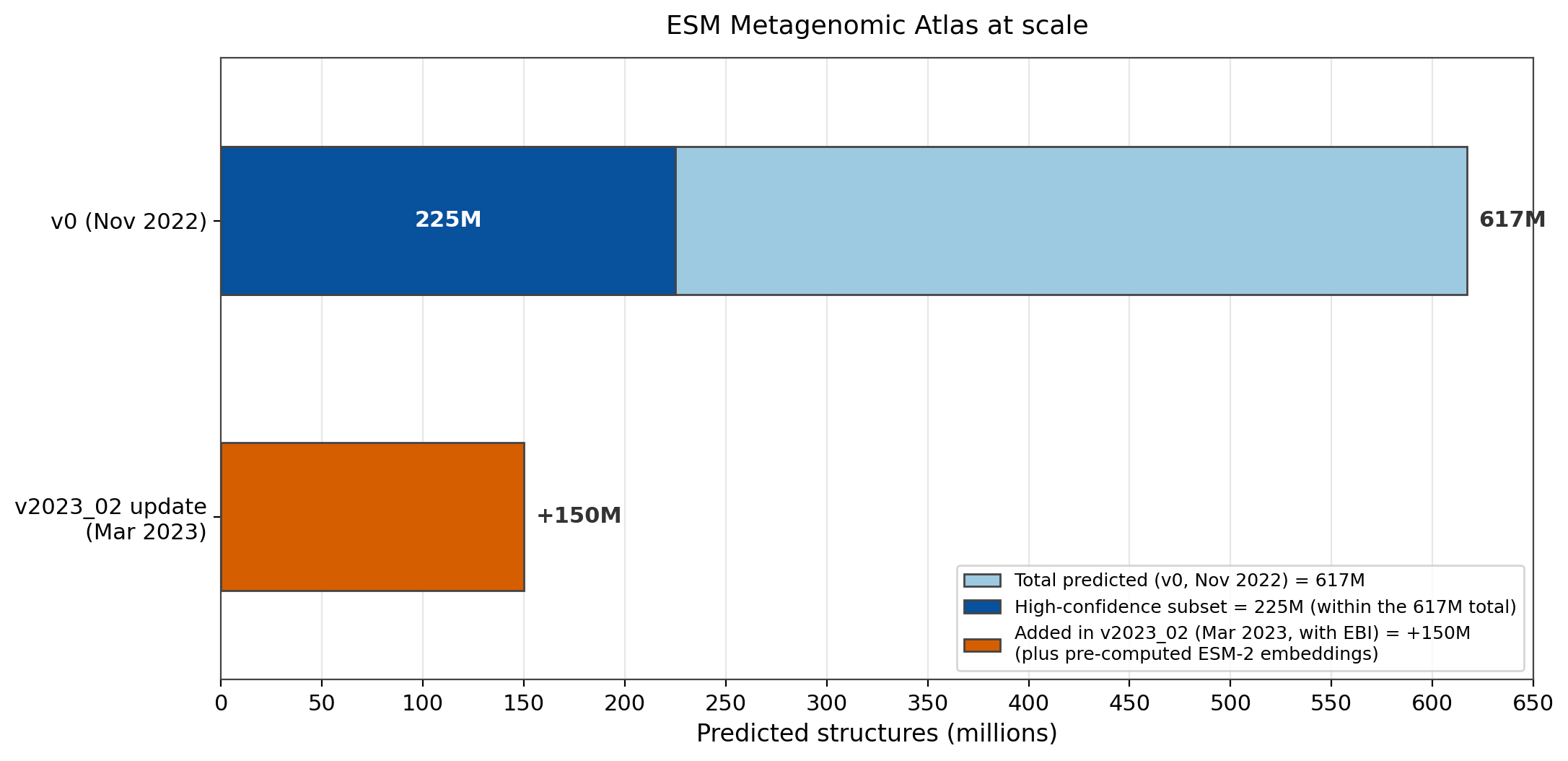 Figure 3: The ESM Metagenomic Atlas holds 617 million predicted structures at the v0 release, 225 million of them high-confidence (shown as a subset of the 617 million total), with a further 150 million added in v2023_02. Source: Lin et al., Science 2023, DOI 10.1126/science.ade2574; ESM GitHub README, facebookresearch/esm.
Figure 3: The ESM Metagenomic Atlas holds 617 million predicted structures at the v0 release, 225 million of them high-confidence (shown as a subset of the 617 million total), with a further 150 million added in v2023_02. Source: Lin et al., Science 2023, DOI 10.1126/science.ade2574; ESM GitHub README, facebookresearch/esm.
These are predictions, not crystals, and the 225 million high-confidence structures are the trustworthy core. But that is a structural view of natural diversity at a scale no MSA-based pipeline could reach in reasonable time. The cost was amortized in the right place: the evolutionary statistics were paid for once during pretraining, so the marginal cost of folding the next sequence is a single forward pass instead of a database search. An MSA-first approach at this scale means hundreds of millions of searches against reference databases; ESMFold trades each one away. Metagenomic sequences come from environmental samples, often from organisms never cultured, and many have no close homolog in any reference database. Those are exactly the sequences where an MSA-based predictor struggles most, because there are too few relatives to align in the first place.
The Atlas is searchable by structure, not only by sequence, which matters because structural homology survives where sequence homology has decayed past detection. Foldseek provides search against the Atlas without the length limitation of the hosted fold tool,36 so a new structure can be queried against hundreds of millions of predicted folds.
What Carries Over
The thread is short. A language model trained only to predict masked residues internalizes structure as it scales.3 A thin folding head reads that structure off frozen embeddings,17 so prediction needs one sequence and no alignment,1 an order of magnitude faster,25 which is what makes 617 million metagenomic structures something you can actually compute.33
The honest caveat is in the benchmark table. At matched evaluation, the gap between single-sequence and MSA-based contact precision has narrowed but not vanished: ESM-2 15B reaches 54.5 on the large-validation set against the MSA Transformer's 57.4.1315 For a well-characterized protein with a deep alignment, the MSA still carries information worth using, and an MSA-based method retains an edge on the hardest targets. The case for ESMFold is the opposite regime: the dark, alignment-poor majority of natural sequences, where speed and MSA independence are not a trade-off but the only way in. Anfinsen said the structure is in the sequence. A large enough language model finally learned to read it there.
References
Footnotes
-
ESMFold model card. ("ESMFold is a state-of-the-art end-to-end protein folding model based on an ESM-2 backbone. It does not require any lookup or MSA step, and therefore does not require any external databases to be present in order to make predictions.") HuggingFace, facebook/esmfold_v1. https://huggingface.co/facebook/esmfold_v1 ↩ ↩2 ↩3 ↩4 ↩5
-
Lin, Z., Akin, H., Rao, R., Hie, B., et al. (2023). Evolutionary-scale prediction of atomic-level protein structure with a language model. Science, 379(6637), 1123-1130. DOI: 10.1126/science.ade2574. ("Recent advances in machine learning have leveraged evolutionary information in multiple sequence alignments to predict protein structure.") https://doi.org/10.1126/science.ade2574 ↩ ↩2 ↩3
-
Lin et al. (2023), Science, DOI: 10.1126/science.ade2574, abstract. ("As language models of protein sequences are scaled up to 15 billion parameters, an atomic-resolution picture of protein structure emerges in the learned representations.") ↩ ↩2 ↩3
-
Editor summary, Baek, M., et al. (2021). Accurate prediction of protein structures and interactions using a three-track neural network. Science. ("In 1972, Anfinsen won a Nobel prize for demonstrating a connection between a protein's amino acid sequence and its three-dimensional structure.") Background context from the RoseTTAFold paper. https://doi.org/10.1126/science.abj8754 ↩
-
Editor summary, Baek, M., et al. (2021), Science. ("Deep learning methods took center stage at CASP14, with DeepMind's Alphafold2 achieving remarkable accuracy.") Background context. https://doi.org/10.1126/science.abj8754 ↩
-
Baek, M., et al. (2021). Accurate prediction of protein structures and interactions using a three-track neural network. Science. They "used a three-track network to process sequence, distance, and coordinate information simultaneously and achieved accuracies approaching those of DeepMind." Background context. https://doi.org/10.1126/science.abj8754 ↩
-
ESM-2 model card. ("ESM-2 is a state-of-the-art protein model trained on a masked language modelling objective.") HuggingFace, facebook/esm2_t33_650M_UR50D. https://huggingface.co/facebook/esm2_t33_650M_UR50D ↩
-
Rives, A., et al. (2021). Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. PNAS, 118(15). DOI: 10.1073/pnas.2016239118. ("We find that without prior knowledge, information emerges in the learned representations on fundamental properties of proteins such as secondary structure, contacts, and biological activity.") ESM-1b, the single-sequence predecessor to ESM-2. https://doi.org/10.1073/pnas.2016239118 ↩
-
Rives, A., et al. (2021), PNAS. ("Unsupervised representation learning... improves state-of-the-art features for long-range contact prediction.") ESM-1b predecessor; 650M parameters, trained on 250 million sequences. https://doi.org/10.1073/pnas.2016239118 ↩
-
ESM GitHub README, facebookresearch/esm. ("For unsupervised contact prediction, a sparse linear combination of the attention heads is used to directly predict protein contacts, fitted with logistic regression on 20 structures.") https://github.com/facebookresearch/esm ↩
-
ESM GitHub README, facebookresearch/esm. ("All contact numbers are the top-L,LR precision metric, where long range means sequence separation of at least 24 residues.") https://github.com/facebookresearch/esm ↩
-
ESM GitHub README comparison table, facebookresearch/esm. ESM-2 unsupervised long-range contact precision (CAMEO column): 8M=15.7, 15B=51.7. (CASP14 column): 8M=9.8, 15B=37.0. https://github.com/facebookresearch/esm ↩
-
ESM GitHub README comparison table, facebookresearch/esm. ESM-2 long-range contact precision, Large-valid column: 8M=15.9, 35M=28.8, 150M=42.2, 650M (700M row)=50.1, 3B=52.7, 15B=54.5. ESM-1b predecessor, Large-valid=41.1. https://github.com/facebookresearch/esm ↩ ↩2
-
ESM GitHub README comparison table, facebookresearch/esm. ESM-2 structure-prediction accuracy, CASP14/CAMEO columns: 8M=36.7/48.1, 35M=41.4/56.4, 150M=49.0/64.9, 650M (700M row)=51.3/70.1, 3B=52.5/71.8, 15B=55.4/72.1. https://github.com/facebookresearch/esm ↩
-
ESM GitHub README comparison table, facebookresearch/esm. ESM-MSA-1b (MSA Transformer), Large-valid contact precision = 57.4; ESM-2 15B, Large-valid contact precision = 54.5. ESM-MSA-1b is an MSA-based model that consumes alignments, not a single-sequence model. https://github.com/facebookresearch/esm ↩ ↩2
-
ESMFold model card. ("ESMFold is a state-of-the-art end-to-end protein folding model based on an ESM-2 backbone.") HuggingFace, facebook/esmfold_v1. https://huggingface.co/facebook/esmfold_v1 ↩
-
ESM GitHub README, facebookresearch/esm. ("For structure prediction, an AlphaFold2 structure module is trained directly from the frozen language model embeddings.") https://github.com/facebookresearch/esm ↩ ↩2 ↩3
-
ESM GitHub README, facebookresearch/esm. ("ESMFold harnesses the ESM-2 language model to generate accurate structure predictions end to end directly from the sequence of a protein.") https://github.com/facebookresearch/esm ↩
-
ESM GitHub README models table, facebookresearch/esm. (esmfold_v1: 48 (+36) layers, 690M (+3B) parameters; 690M folding head on the 3B ESM-2 trunk.) https://github.com/facebookresearch/esm ↩
-
ESM GitHub README, facebookresearch/esm. ("Besides esm.pretrained.esmfold_v1() which is the best performing model we recommend using, we also provide esm.pretrained.esmfold_v0() which was used for the experiments in Lin et al. 2022.") Earlier versions appeared as preprint bioRxiv 2022.07.20.500902. https://github.com/facebookresearch/esm ↩
-
ESM GitHub README, facebookresearch/esm. ("Number of recycles to run. Defaults to number used in training (4).") https://github.com/facebookresearch/esm ↩
-
ESM GitHub README, facebookresearch/esm. ("Chunks axial attention computation to reduce memory usage from O(L^2) to O(L).") https://github.com/facebookresearch/esm ↩
-
ESM GitHub README, facebookresearch/esm. Per-residue confidence reported as pLDDT, stored in the B-factor column ("# this will be the pLDDT" on the mean B-factor). https://github.com/facebookresearch/esm ↩ ↩2 ↩3
-
ESM GitHub README, ESMStructuralSplitDataset, facebookresearch/esm. ("The dataset implements structural holdouts at the family, superfamily, and fold level. The SCOPe database is used to classify domains.") https://github.com/facebookresearch/esm ↩
-
Lin et al. (2023), Science, DOI: 10.1126/science.ade2574, abstract. ("This results in an order-of-magnitude acceleration of high-resolution structure prediction, which enables large-scale structural characterization of metagenomic proteins.") https://doi.org/10.1126/science.ade2574 ↩ ↩2 ↩3 ↩4
-
ESMFold model card. ("As a result, inference time is very significantly faster than AlphaFold2.") HuggingFace, facebook/esmfold_v1. https://huggingface.co/facebook/esmfold_v1 ↩
-
ESM GitHub README, facebookresearch/esm. ("We also provide an API which you can access through curl or on the ESM Metagenomic Atlas web page.") Endpoint: https://api.esmatlas.com/foldSequence/v1/pdb/ . https://github.com/facebookresearch/esm ↩
-
ESM GitHub README, facebookresearch/esm. ("An easy way to get started is to load ESM or ESMFold through the HuggingFace transformers library.") https://github.com/facebookresearch/esm ↩
-
ESM GitHub README, facebookresearch/esm. ("To use the ESMFold model, make sure you start from an environment with python <= 3.9 and pytorch installed.") https://github.com/facebookresearch/esm ↩
-
ESM-2 model card. ("It is suitable for fine-tuning on a wide range of tasks that take protein sequences as input.") HuggingFace, facebook/esm2_t33_650M_UR50D. https://huggingface.co/facebook/esm2_t33_650M_UR50D ↩
-
ESM GitHub README, facebookresearch/esm. ("Note that ESM-2 could be used for variant prediction as well, and is expected to have similar performance to ESM-1v.") https://github.com/facebookresearch/esm ↩
-
ESM GitHub README, facebookresearch/esm. 15B inference can hit OOM on a single GPU; Fairscale Fully Sharded Data Parallel (FSDP) with CPU offloading "allows to do inference of large models on a single GPU." https://github.com/facebookresearch/esm ↩
-
Lin et al. (2023), Science, DOI: 10.1126/science.ade2574, abstract. ("We apply this capability to construct the ESM Metagenomic Atlas by predicting structures for >617 million metagenomic protein sequences, including >225 million that are predicted with high confidence.") https://doi.org/10.1126/science.ade2574 ↩ ↩2
-
ESM GitHub README, facebookresearch/esm. ("In November 2022, we released v0 of the ESM Metagenomic Atlas, an open atlas of 617 million predicted metagenomic protein structures.") https://esmatlas.com ↩
-
ESM GitHub README, facebookresearch/esm. ("The Atlas was updated in March 2023 in collaboration with EBI. The new v2023_02 adds another 150 million predicted structures to the Atlas, as well as pre-computed ESM2 embeddings.") https://github.com/facebookresearch/esm ↩
-
ESM GitHub README, facebookresearch/esm. ("Foldseek provides search against the Atlas without the length limitation.") https://search.foldseek.com/search ↩