The hardest loop in an antibody is CDR-H3, and after five years of antibody-specific foundation models, the best specialized structure predictors still land it at roughly 3 Angstroms of backbone RMSD. IgFold puts the loop at about 3.27 Angstroms.1 ABodyBuilder developers report CDR-H3 as the least accurate region despite improvements.2 tFold-Ab reports 3.01 Angstroms.3 BALMFold reports 3.05.4 These are good numbers. They are also a wall. Every model on this list was trained, tuned, and benchmarked against that loop, and none has knocked it down to the sub-Angstrom accuracy they routinely hit on antibody framework regions.
That gap is half the story. The other half is the license file. Five of the ten leading antibody-specific models lack a clean permissive license for commercial use: four carry explicitly non-commercial terms and one ships weights under unconfirmed terms,54163 which for a field whose output is eventually a drug candidate is not a footnote you can sort out later. Keep two ledgers while reading this literature. The capability ledger records what a model can demonstrably do on a held-out test set: sequence recovery, embedding quality, structure RMSD, inference speed. The validity ledger records what survives contact with a real campaign: a candidate that expresses, folds, binds the intended antigen, clears developability filters, and is legally yours to ship. Antibody foundation models are sold almost entirely on the first ledger. The therapeutic decision lives almost entirely on the second.
This is a survey of the ten antibody-specific foundation models in the rewire.it "Foundation Models in Biology" series: AbLang/AbLang2, ABodyBuilder2/ABodyBuilder3, AntiBERTa/AntiBERTa2, AntiBERTy, BALM, IgBERT/IgT5, IgFold, IgLM, p-IgGen, and tFold-Ab. I organize them so you can decide which to use by task, hardware, and license rather than by leaderboard, and I keep the two ledgers separate on purpose.

Why antibodies need their own models
A reasonable first question: why not just use ESM-2 or AlphaFold? Antibodies are proteins, and the general models are bigger and trained on more data. The answer is in the data and the geometry, and it comes down to three structural facts that break the general-model assumption hard enough to justify a separate field.
The function lives in the loops, not the fold. An antibody variable region is a conserved immunoglobulin scaffold with six hypervariable loops grafted onto it: three complementarity-determining regions (CDRs) on the heavy chain, three on the light. Binding specificity concentrates in those loops, and disproportionately in CDR-H3, the longest and most variable, formed across the V(D)J recombination junction. A general model trained to get the overall fold right is rewarded for nailing the scaffold, which is easy, and barely penalized for missing the loops, which is the part you care about. The ABodyBuilder developers state it plainly: CDR-H3 remains the least accurate region despite improvements.2 Every structure model here reports CDR-H3 RMSD as its headline difficulty metric because that is the region general folders get wrong.
The chains come in pairs. A functional binding site is formed by a heavy and a light chain together, and the paratope draws residues from both. A model that embeds a heavy chain in isolation has discarded half the context before it starts. The newer models, AbLang2,7 IgBERT/IgT5,8 and p-IgGen,9 make native heavy-light pairing a design target. Where a model trains on chains independently, as the base BALM does, it cannot natively model that pairing, a limitation its authors flag directly.4
The data is a different database. General protein models learn from UniProt-style sequence space (UniRef, BFD). Antibodies are underrepresented there, and the interesting variation is somatic, generated within an individual's B cells, not captured by evolutionary databases. The relevant corpus is the Observed Antibody Space (OAS), a collection of repertoire sequences that runs into the billions. AntiBERTy trained on 558 million OAS sequences,10 AntiBERTa2 on roughly 779.4 million,5 BALM on about 336 million clustered to 40 percent non-redundancy,4 and IgBERT/IgT5 on more than 2 billion unpaired sequences plus around 2 million natively paired chains.8 This is a different training distribution, enriched for the framework-plus-hypervariable-loop architecture and the somatic hypermutation of affinity maturation, and it is the one that matters for antibodies.
Those three differences, the loops, the pairing, and the database, are the axes every model below sits on.
The ten models, in one table
Here is the full field on one screen. Read the License column first.
| Model | Function | Developer | Released | Params | License | Commercial? |
|---|---|---|---|---|---|---|
| AntiBERTy | Embeddings | Gray Lab, JHU | 202110 | ~26M10 | MIT10 | Yes |
| IgLM | Generation | Gray Lab, JHU | 20216 | ~13M6 | JHU Academic6 | No |
| AntiBERTa / AntiBERTa2 | Embeddings | Alchemab + OPIG | 2021 / 2023511 | 86M / 202M5 | Non-commercial5 | No |
| AbLang / AbLang2 | Embeddings | OPIG, Oxford | 2022 / 2024127 | Undisclosed12 | BSD 3-Clause12 | Yes |
| IgFold | Structure | Gray Lab, JHU | 20221 | Undisclosed1 | JHU Academic1 | No |
| tFold-Ab | Structure | Tencent AI Lab | 20223 | 650M PLM; trunk undisclosed3 | PolyForm Noncommercial3 | No |
| ABodyBuilder2 / 3 | Structure | OPIG / Exscientia | 2022 / 2024132 | Undisclosed2 | BSD / Apache-2.0132 | Yes |
| BALM / BALMFold | Embeddings + Structure | BEAM-Labs, Fudan | 20234 | ~150M4 | MIT code; weights unclear4 | Unconfirmed |
| IgBERT / IgT5 | Embeddings | Exscientia + Oxford | 20248 | 0.4B / 3B8 | MIT8 | Yes |
| p-IgGen | Generation | OPIG + AstraZeneca | 20249 | ~17.35M9 | BSD 3-Clause9 | Yes |
Table 1: The ten antibody-specific foundation models. "Undisclosed" means the paper or repository does not state an exact parameter count. BALMFold is BALM's structure derivative, counted with the BALM family. Sources cited per cell.12721351110481693
Two patterns jump out before any benchmark. First, the models are small. IgLM is about 13 million parameters,6 p-IgGen about 17.35 million,9 AntiBERTy about 26 million.10 Even the largest, IgT5 at 3 billion,8 is dwarfed by general genomic or protein models. Antibody modeling has not needed scale the way general protein modeling has, because the search space is narrower: a conserved scaffold with hypervariable loops, captured in repertoire databases that already number in the billions. Parameter count is a poor proxy for usefulness here; p-IgGen, two orders of magnitude smaller than IgT5, posts the best zero-shot immunogenicity correlation in its paper.9
Second, the license column does not track quality, and the developer concentration explains why. The Oxford Protein Informatics Group (OPIG) and the Johns Hopkins Gray Lab between them account for six of the ten, and the licenses split by lab: OPIG and Exscientia ship permissive, commercially usable models, while Johns Hopkins and Tencent ship research-only. Hold that thought.
Three functions, not one
The ten models do three different jobs, and conflating them is the most common mistake practitioners make. A model that produces excellent embeddings does not predict structure. A model that generates sequences does not score binding affinity. Match the model to the function.

Function one: embeddings and representation
Most of the list lives here. These are encoder language models trained with masked language modeling on antibody repertoires, producing per-residue and per-sequence vectors you feed to downstream predictors. They differ in scale, in whether they handle paired chains, and in the bias each was built to correct.
AntiBERTy is the lightweight workhorse: a BERT-style encoder, 8 layers, 8 heads, 512 hidden dimensions, about 26 million parameters, trained on 558 million OAS sequences.10 It generates embeddings, classifies species and chain type, fills masked residues, and computes pseudo-log-likelihood naturalness scores.10 Its primary paper offers demonstrations rather than leaderboard numbers, which is honest: it clusters repertoires into trajectories resembling affinity maturation and, under weakly supervised learning, recovers key binding residues.10 Its real significance is as infrastructure. AntiBERTy is the language-model component inside IgFold,10 which means an MIT-licensed embedding model underpins a non-commercially-licensed structure tool. Its limitation is plainly stated: small, sequence-only, no structure or antigen context.10
AbLang and AbLang2 come from OPIG, with separate heavy and light chain models in v1 (a RoBERTa-style encoder, 12 blocks, 12 heads, 768 hidden) trained on roughly 14.1 million heavy and 187,000 light chain OAS sequences.12 AbLang's original selling point was restoration: it restores missing antibody residues, notably the frequently-missing first ~15 N-terminal residues in OAS, better than IMGT germline imputation and better than the general protein model ESM-1b, without needing germline knowledge, and about 7x faster than ESM-1b.12 AbLang2 was built specifically to fix a flaw in v1, germline bias, which over-predicted germline residues and hurt exactly when you care about the somatic mutations that distinguish a matured antibody; it uses focal loss to mitigate that bias and adds paired heavy-light modeling.7 The license is permissive BSD 3-Clause.12 This is honest iteration: the v2 paper names the v1 failure mode and addresses it.
AntiBERTa and AntiBERTa2 from Alchemab scaled the idea up: v1 at 86 million parameters, v2 at 202 million.5 AntiBERTa v1 reported state-of-the-art paratope prediction at publication, beating prior tools like Parapred and ProABC-2.5 The CSSP variant adds a contrastive sequence-structure objective.11 The catch is the license: AntiBERTa2 is non-commercial only, with even generated sequences restricted to non-commercial use, and commercial licensing routed through Alchemab.5 If you are in industry, this is a benchmark to beat, not a tool to use.
IgBERT and IgT5 from Exscientia are the paired-chain specialists and the most rigorous benchmark in the set, fine-tuned from ProtBert and ProtT5 on more than 2 billion unpaired plus about 2 million natively paired OAS sequences.8 They are the cleanest demonstration of the antibody-specific premium: on 15% masked sequence recovery, IgT5 achieves the highest recovery across nearly all CDRs, and the antibody-specific models substantially beat general protein language models in the hypervariable loops H3 and L3.8 The paper's honesty is its strongest feature: on expression prediction, the general ProtT5 had the highest R-squared, and the antibody-specific models did not win.8 That single negative result is worth more than a dozen leaderboard wins, because it tells you where antibody pretraining stops helping. MIT licensed, commercially usable.8
BALM from BEAM-Labs takes a different architectural route: a 30-block encoder initialized from the ESM-2 150M checkpoint, with an antibody-specific positional embedding based on IMGT numbering replacing the standard scheme, plus an adaptive entropy-based masking strategy.4 Trained on about 336 million OAS sequences, it reports state-of-the-art results across four antigen-binding and function tasks.4 One practical warning: at least three unrelated tools are called "BALM," including a protein-ligand binding-affinity model and a separate brineylab "BALM-paired" project, so cite the right one.4
Function two: Fv structure prediction
Three antibody-specific structure predictors compete, plus BALM's BALMFold derivative. They share one move: drop AlphaFold's MSA-dependent machinery, which is slow and ill-suited to antibodies whose diversity comes from recombination rather than evolution, and replace it with a single-sequence language model front end. The payoff is speed measured in seconds rather than minutes, at accuracy that is competitive on framework regions and still imperfect on CDR-H3.
IgFold pairs AntiBERTy's 512-dimensional embeddings with graph-transformer and invariant-point-attention layers, then refines with Rosetta, no MSA required.1 It established the speed argument: framework backbone RMSD around 0.46 to 0.48 Angstroms versus about 0.66 to 0.69 for AlphaFold-Multimer, CDR-H3 around 3.27 Angstroms versus about 3.56, and a full structure in under 25 seconds versus over 7 minutes for AlphaFold-Multimer.1 Roughly comparable accuracy, an order of magnitude faster. The license is the gate: JHU Academic Software License, non-commercial, with commercial use requiring a separate agreement through Johns Hopkins Tech Ventures.1
ABodyBuilder2 and ABodyBuilder3 are the OPIG and Exscientia line, derived from the AlphaFold-Multimer structure module.2 ABodyBuilder2, part of the ImmuneBuilder suite, is an ensemble of four models using ensemble disagreement for confidence, reported as competitive with or better than AlphaFold-Multimer and IgFold on CDR-H3 while being more than 100x faster.13 ABodyBuilder3 swaps one-hot encoding for ProtT5 embeddings, replaces the ensemble with a single model plus a predicted-lDDT confidence head, and reports state-of-the-art CDR loop accuracy with reduced CDR-H3 and CDR-L3 backbone RMSD.2 It is the permissive choice: ABodyBuilder2 is BSD 3-Clause, ABodyBuilder3 code is Apache-2.0, both commercially usable.132 One operational caveat for the LM variant: it depends on an external ~3B-parameter ProtT5 encoder, which adds compute and memory.2
tFold-Ab from Tencent replaces AlphaFold-Multimer's MSA-dependent Evoformer with embeddings from ESM-PPI, a 650-million-parameter language model derived from ESM2-650M and further pretrained on UniRef50, PDB, protein-protein interaction data, and antibody data.3 It claims roughly 1000x speedup over AlphaFold-Multimer.3 On the held-out SAbDab-22H2 antibody set it reports CDR-H3 RMSD 3.01 Angstroms and DockQ 0.770, against AlphaFold-Multimer at 3.07 / 0.773, Chai-1 at 3.25 / 0.772, IgFold at 3.37 / 0.715, and ImmuneBuilder at 3.46 / 0.749.3 Read those margins carefully: tFold-Ab edges AlphaFold-Multimer on CDR-H3 RMSD by 0.06 Angstrom and trails it by 0.003 on DockQ. This is a tie dressed as a win, a cluster of methods landing near the same wall, and that wall is CDR-H3. The license is the constraint: PolyForm Noncommercial 1.0.0.3
BALMFold, the structure derivative of BALM, reports paired CDR-H3 RMSD of 3.05 Angstroms against AlphaFold2-Multimer's 3.56, IgFold's 3.42, OmegaFold's 3.55, and ESMFold's 4.41, with inference around 5 seconds per structure, roughly 6x faster than IgFold.4 It carries BALM's weights-license ambiguity.4

Function three: sequence generation
Two decoder models generate antibody sequences, and both are surprisingly tiny, which is itself the story.
IgLM is the original: a GPT-2-style decoder, about 13 million parameters, trained with a text-infilling objective on 558 million unpaired OAS sequences across six species (human, mouse, rat, rabbit, rhesus macaque, camel), conditioned on chain type and species control tokens.6 The infilling objective is the clever part: it lets you mask a variable-length span, typically a CDR loop, and regenerate it with bidirectional context, exactly the antibody-engineering operation you want. Its reported wins are in silico: infilled CDR loops achieved lower perplexity than ProGen2-OAS, and infilling-based redesign improved developability and humanness scores.6 Two honest caveats: it trains on unpaired sequences only, and those developability and humanness gains are in silico, not experimentally validated in the core paper.6 The license is JHU Academic, non-commercial.6
p-IgGen from OPIG and AstraZeneca is the paired generator: a small 17.35-million-parameter GPT-2-style decoder with rotary embeddings, trained in stages on human OAS, roughly 250 million unpaired sequences then around 1.8 million paired chains, with a "developable" variant biased toward TAP developability criteria.9 By concatenating light chain to heavy chain with random forward/reverse presentation, it generates a VH given a VL or vice versa.9 Its headline result punches above its size: zero-shot immunogenicity prediction at Pearson r=0.53, ahead of IgLM at 0.20 and AntiBERTy at -0.05.9 On expression it beat the antibody-specific IgLM and AntiBERTy but underperformed the much larger general model ProGen, the same lesson IgBERT taught.9 p-IgGen is BSD 3-Clause and fully commercial.9
If you want to load one of these and start producing embeddings, the permissive embedding models are a two-line job from Hugging Face. IgBERT is a useful default because it gives paired-chain awareness at a manageable 400M:
"""
igbert_embeddings.py
Load IgBert (Exscientia/IgBert) and embed a paired heavy + light antibody.
License note: IgBert and its larger ~3B sibling IgT5 (Exscientia/IgT5) are
released under the MIT license. IgT5 loads the same way via its T5 encoder
(T5EncoderModel.from_pretrained("Exscientia/IgT5")) and exposes the same
last_hidden_state interface, just with a wider embedding dimension.
Output shapes (IgBert, hidden size 1024):
- per_residue : (num_tokens, 1024) one vector per residue (+ special tokens)
- sequence : (1024,) mean-pooled over real residues
"""
import torch
from transformers import BertModel, BertTokenizer
# Per the model card, residues are space-separated and the heavy chain comes
# first, then the light chain, joined by a " [SEP] " token.
heavy = "QVQLVQSGAEVKKPGASVKVSCKASGYTFTSYAMHWVRQAPGQRLEWMGWINAGNGNTKYSQKFQG"
light = "DIQMTQSPSSLSASVGDRVTITCRASQSISSYLNWYQQKPGKAPKLLIYAASSLQSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYC"
paired = " ".join(heavy) + " [SEP] " + " ".join(light)
tokenizer = BertTokenizer.from_pretrained("Exscientia/IgBert", do_lower_case=False)
model = BertModel.from_pretrained("Exscientia/IgBert", add_pooling_layer=False)
model.eval()
tokens = tokenizer(paired, return_tensors="pt")
with torch.no_grad():
output = model(**tokens)
# Per-residue embeddings: (batch, num_tokens, 1024) -> drop the batch dim.
per_residue = output.last_hidden_state.squeeze(0)
# Sequence-level embedding: mean-pool over real tokens using the attention mask.
mask = tokens["attention_mask"].squeeze(0).unsqueeze(-1)
sequence = (per_residue * mask).sum(0) / mask.sum()
print("per-residue embedding shape:", tuple(per_residue.shape))
print("sequence embedding shape: ", tuple(sequence.shape))
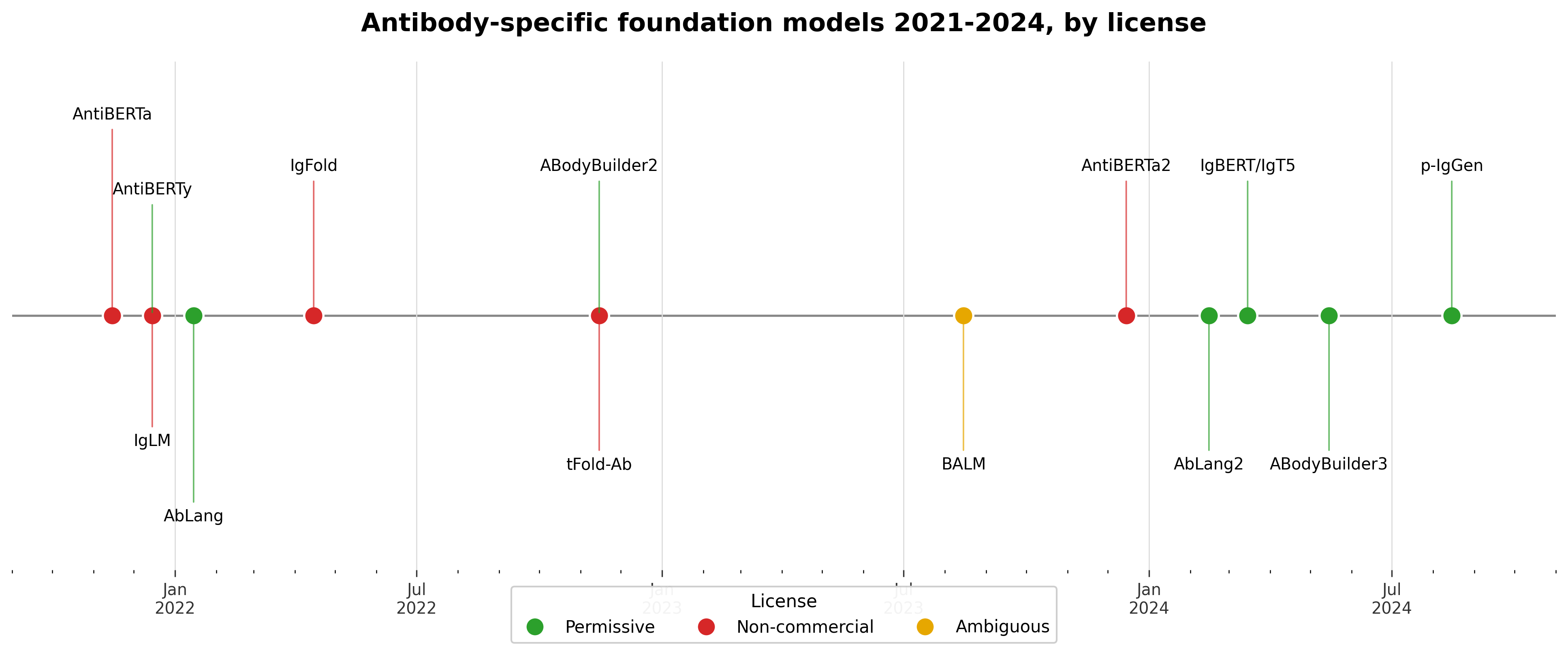
The timeline tells a story the table flattens. The non-commercial models cluster in the early period, 2021 and 2022: IgLM, IgFold, AntiBERTa, tFold-Ab.6153 The clearly permissive, paired-chain, commercially-deployable models, AbLang2, IgBERT/IgT5, ABodyBuilder3, p-IgGen, all arrive in 2024.7829 If you are starting a commercial program today, the field has moved in your favor: the strongest recent releases are also the ones you can actually use.
The capability ledger versus the validity ledger
Now the separation that matters. Here is what the structure models score, in silico, on held-out benchmarks. These are real, comparable within their frame, and impressive.
| Model | CDR-H3 RMSD (paired Ab) | Speed claim | Commercial? | Source |
|---|---|---|---|---|
| tFold-Ab | 3.01 A | ~1000x vs AF-Multimer | No (PolyForm NC) | 3 |
| BALMFold | 3.05 A | ~5 s/structure, ~6x vs IgFold | Unconfirmed | 4 |
| IgFold | ~3.27 A | <25 s vs >7 min | No (JHU) | 1 |
| ABodyBuilder2 | competitive w/ AF-Multimer | >100x faster | Yes (BSD) | 13 |
| AlphaFold-Multimer (baseline) | 3.07 A | minutes | n/a | 3 |
Table 2: The capability ledger for structure prediction. CDR-H3 figures come from each model's own paper on its own test split, except where a single common benchmark (tFold-Ab's SAbDab-22H2) places several methods together; the column is indicative, not a strict head-to-head. Sources cited per row.13413
Every model clears the bar it set for itself. Now the validity ledger, which the same papers are honest about when you read past the abstract.
| In-silico capability (capability ledger) | What the validity ledger records |
|---|---|
| CDR-H3 RMSD ~3 A across all structure models134 | CDR-H3 remains the least accurate region; ~3 A is too coarse to trust loop conformation for design214 |
| IgT5 wins sequence recovery across nearly all CDRs8 | General ProtT5 won expression prediction; antibody pretraining does not help all tasks8 |
| IgLM improves developability and humanness scores6 | Those gains are in silico, not experimentally validated in the core paper6 |
| p-IgGen scores immunogenicity at r=0.539 | A correlation, not a clearance; no antigen conditioning, no binding-affinity prediction9 |
| tFold-Ag models antibody-antigen complexes14 | DockQ 0.217, success 28.3%, below AlphaFold-3's 0.257 / 32.3%; "still-hard nature of antibody-antigen docking"14 |
Table 3: Capability versus validity. The two ledgers diverge most as you approach the therapeutic question. Sources cited per row.2869314
The pattern is consistent. The closer you get to the therapeutic question, the larger the gap. Sequence recovery is a solved-ish problem. Fv framework structure is a solved-ish problem. CDR-H3 structure is not, and it is precisely the region that determines binding. Antibody-antigen docking, the thing de-novo discovery actually needs, sits at DockQ 0.217 and a 28.3% success rate even on the strongest specialized model, below AlphaFold-3.14
This is why "improved in-silico developability" should be read as a hypothesis, not a result. A benchmark number is a hypothesis about real-world performance, not a measurement of it. The generative models do not condition on a target antigen and do not predict binding affinity to it.9 They produce antibody-like sequences with good naturalness scores. Whether those sequences bind anything is a separate, unvalidated question that the wet lab answers.
What the benchmarks do not tell you
A survey that only reports wins is a marketing document. Three gaps deserve to stay in front of you, and being explicit about the holes is part of the job.
Parameter counts are frequently undisclosed. AbLang, ABodyBuilder2/3, IgFold's structure trunk, and tFold-Ab's folding trunk do not publish exact parameter counts.12213 When a paper or model card does not state a number, this survey says "undisclosed" rather than guess. What is disclosed for ABodyBuilder is structural: v2 is an ensemble of four AlphaFold-style models, v3 a single model.2
Benchmarks are mostly author-reported on bespoke splits. Almost every accuracy figure here comes from the model's own paper on its own test set. There is no single shared, independently administered antibody structure leaderboard the way CASP serves general structure prediction. The cross-model tables in the tFold3 and BALM4 papers are more useful than single-model claims because they evaluate several methods on a common held-out set, but they still carry the incumbent's framing and choice of baselines. tFold-Ab's comparisons against Chai-1 and AlphaFold-3 were added to the repository later.14 Before you trust any of these on your data, beat a trivial baseline first.
In silico is not in vitro. This is the gap that swallows the others. IgLM's developability and humanness improvements were in silico and, in the core paper, not experimentally validated.6 p-IgGen's developability biasing is heuristic TAP-based filtering, not a manufacturability guarantee.9 None of these papers reports a de-novo wet-lab binding hit rate from sequences the model designed unconditioned on antigen. Generative antibody campaigns in practice still see wet-lab hit rates on the order of a percent, and no model here changes that arithmetic by itself. What these models do is enrich: they raise the fraction of designs worth synthesizing, shrink libraries, and score candidates before you spend bench time. That is genuinely valuable. It is not the same as designing a binder, and treating a low perplexity or a good pLDDT as a binding prediction is the most common way to misuse them.
One more hole worth naming: BALM's weights license is unconfirmed. The code is MIT, but the weights ship via a Google Drive link without an explicit standalone license, and the journal article itself is published under a CC BY-NC (non-commercial) Creative Commons license that governs the text rather than the weights.4 Treat commercial use of the weights as unverified until you confirm it in writing.
License is a first-class deployment constraint
For an industry practitioner, license is not an appendix, it is the first filter. Permissive licenses (MIT, BSD, Apache-2.0) allow commercial use with attribution. Research-only licenses let you download weights and publish, but block commercial deployment without a separately negotiated agreement. The models cluster into three tiers.
| Tier | Models | License | What it means for commercial use |
|---|---|---|---|
| Permissive (ship it) | AntiBERTy10, AbLang/AbLang212, IgBERT/IgT58, p-IgGen9, ABodyBuilder2/3132 | MIT, BSD 3-Clause, Apache-2.0 | Commercial use permitted, attribution only. No separate agreement needed. |
| Encumbered (negotiate or walk) | AntiBERTa/AntiBERTa25, IgFold1, IgLM6, tFold-Ab3 | Alchemab non-commercial; JHU Academic Software License; PolyForm Noncommercial 1.0.0 | Weights are downloadable, but use is research-only. Commercial deployment requires a separately executed agreement with Alchemab, Johns Hopkins Tech Ventures, or Tencent. |
| Ambiguous (verify before you build) | BALM4 | MIT code; weights via Google Drive with no explicit weights license | Code permits commercial use; the weights carry no formal license, so their commercial status is unconfirmed. Verify directly. |
Table 4: The openness matrix. "Downloadable" is not "open." Sources cited per row.12213510481693
The trap is in the middle tier: the weights are openly downloadable. You can install IgFold and IgLM, run them, get results, and never see a license prompt.16 The JHU Academic Software License grants a non-exclusive, non-transferable license for non-commercial purposes only, including non-commercial research conducted at commercial entities; commercial use requires a separately executed written agreement.16 AntiBERTa2's weights sit on Hugging Face under a license tag of "other," with the model card stating that both the models and the antibody sequences they generate are for non-commercial use only.5 tFold-Ab is governed by PolyForm Noncommercial 1.0.0, openly downloadable weights under explicitly non-commercial terms.3 Downloading any of these into a commercial pipeline is a license violation, not a gray area.
The practical consequence: if you are building a commercial pipeline, your structure-prediction default is ABodyBuilder3 (Apache-2.0)2 rather than the otherwise-comparable tFold-Ab (PolyForm Noncommercial)3 or IgFold (JHU non-commercial).1 Your embedding default is IgBERT/IgT5 or AbLang2, both permissive.87 Your generative default is p-IgGen (BSD)9 rather than IgLM (JHU non-commercial).6 The accuracy differences between the permissive and restricted options in each category are small enough that the license usually decides. And because the permissive cluster spans all three functions, you can build a fully commercial-clean pipeline, embed with AbLang2 or IgBERT, fold with ABodyBuilder3, generate with p-IgGen, without a single non-commercial dependency.
How to choose, by task
Decide by task and license, not by leaderboard. One orientation note first: antibody numbering and CDR annotation (IMGT, Kabat, Chothia) are not foundation-model tasks. They are deterministic or HMM-based preprocessing steps handled by tools like ANARCI that feed these models rather than competing with them, so do not reach for a 3B-parameter model to do an HMM's job. BALM is the only model here that folds numbering into its architecture, via IMGT positional embeddings.4
- Need fast embeddings for a property predictor, commercial OK? AntiBERTy for a lightweight MIT-licensed backbone,10 or IgBERT/IgT5 if you want paired-chain awareness and the strongest CDR sequence recovery.8 Benchmark against a general ProtT5 baseline first, because for some tasks like expression prediction the general model wins.8
- Need paired heavy-light representation? AbLang2,7 IgBERT/IgT5,8 or p-IgGen.9 Skip base BALM, which does not model native pairing.4
- Need germline-aware representation or residue restoration? AbLang2, BSD-licensed, built specifically to correct germline bias.7
- Need Fv structure, commercial OK? ABodyBuilder3, Apache-2.0, single-model with a calibrated pLDDT head.2 Expect about 3 Angstroms on CDR-H3 and treat that loop's conformation as approximate.2
- Need Fv structure, research only, want the published accuracy edge? tFold-Ab edges AlphaFold-Multimer on CDR-H3 RMSD,3 but read the PolyForm Noncommercial terms before you build on it.3 IgFold is the well-understood fast baseline, also non-commercial.1
- Need to generate or redesign CDR loops? IgLM for infilling (non-commercial),6 p-IgGen for paired de-novo generation (BSD, commercial OK).9 In both cases the sequences are antibody-like by construction; binding to your antigen is an empirical question they do not answer.9
- Need antibody-antigen complex structure for de-novo discovery? Manage expectations. The best specialized result is DockQ 0.217 at 28.3% success, below AlphaFold-3, on a still-hard problem.14
The bottom line
Antibody-specific foundation models earned their place. They beat general protein models in the hypervariable loops,8 they predict Fv structure in seconds instead of minutes,1 and they encode real biology that ESM-2 ignores: OAS repertoire distributions, CDR geometry, heavy-light pairing. On the capability ledger, the field has delivered.
The validity ledger is where the work remains. CDR-H3, the loop that decides binding, sits at a 3-Angstrom wall that five years and ten models have not broken.2134 Antibody-antigen docking, the function de-novo discovery actually needs, sits below AlphaFold-3 at under 30% success.14 The developability and immunogenicity wins are in-silico correlations awaiting wet-lab confirmation.69 None of this makes the models useless. It makes them tools for a specific stage of the pipeline, representation and fast structural triage, rather than a replacement for the bench.
So read every benchmark number as a hypothesis about the validity ledger, keep the two ledgers separate, and pick by task and license. In a field where the output is a drug candidate, the license is the first filter, not the last; start your shortlist from the openness matrix, then optimize for accuracy within the tier you are allowed to ship. Doing it the other way around is how a program discovers, in legal review, that its favorite model was never an option. The models that win the capability ledger and the license check, ABodyBuilder3, IgBERT/IgT5, AbLang2, p-IgGen, are the ones you can actually deploy.
References
Footnotes
-
Ruffolo, Chu, Mahajan, Gray. "Fast, accurate antibody structure prediction from deep learning on massive set of natural antibodies" (IgFold). Nature Communications 14, 2389, 2023. https://www.nature.com/articles/s41467-023-38063-x ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20 ↩21 ↩22
-
Kenlay H., Dreyer F.A., Cutting D., Nissley D., Deane C.M. "ABodyBuilder3: improved and scalable antibody structure predictions." Bioinformatics 40(10):btae576, 2024. https://doi.org/10.1093/bioinformatics/btae576 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20 ↩21
-
Wu J., Wu F., Jiang B., Liu W., Zhao P. "tFold-Ab: Fast and Accurate Antibody Structure Prediction without Sequence Homologs." bioRxiv 2022.11.10.515918, 2022. https://www.biorxiv.org/content/10.1101/2022.11.10.515918v1 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20 ↩21 ↩22 ↩23 ↩24 ↩25
-
"Accurate prediction of antibody function and structure using bio-inspired antibody language model" (BALM / BALMFold). Briefings in Bioinformatics 25(4):bbae245, 2024. https://doi.org/10.1093/bib/bbae245 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20 ↩21 ↩22 ↩23
-
Leem et al. "Deciphering the language of antibodies using self-supervised learning" (AntiBERTa). Patterns (Cell Press), 2022. https://www.cell.com/patterns/fulltext/S2666-3899(22)00105-2 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13
-
Shuai, Ruffolo, Gray. "IgLM: Infilling language modeling for antibody sequence design." Cell Systems 14(11):979-989.e4, 2023. https://doi.org/10.1016/j.cels.2023.10.001 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20 ↩21 ↩22
-
Olsen, Moal, Deane. "Addressing the antibody germline bias and its effect on language models for improved antibody design" (AbLang2). bioRxiv 2024.02.02.578678, 2024. https://doi.org/10.1101/2024.02.02.578678 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8
-
Kenlay et al. "Large scale paired antibody language models" (IgBERT / IgT5). PLOS Computational Biology, 2024. https://doi.org/10.1371/journal.pcbi.1012646 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20 ↩21 ↩22
-
Turnbull OM, Oglic D, Croasdale-Wood R, Deane CM. "p-IgGen: a paired antibody generative language model." Bioinformatics 40(11):btae659, 2024. https://doi.org/10.1093/bioinformatics/btae659 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20 ↩21 ↩22 ↩23 ↩24 ↩25
-
Ruffolo, Gray, Sulam. "Deciphering antibody affinity maturation with language models and weakly supervised learning" (AntiBERTy). arXiv:2112.07782, 2021. https://arxiv.org/abs/2112.07782 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14
-
Barton et al. "Enhancing Antibody Language Models with Structural Information" (AntiBERTa2-CSSP). NeurIPS 2023 MLSB / bioRxiv 2023.12.12.569610, 2023. https://www.biorxiv.org/content/10.1101/2023.12.12.569610v1 ↩ ↩2 ↩3
-
Olsen, Moal, Deane. "AbLang: an antibody language model for completing antibody sequences." Bioinformatics Advances 2(1):vbac046, 2022. https://doi.org/10.1093/bioadv/vbac046 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10
-
Abanades B. et al. "ImmuneBuilder: Deep-Learning models for predicting the structures of immune proteins" (introduces ABodyBuilder2). Communications Biology, 2023; bioRxiv 2022.11.04.514231. https://doi.org/10.1101/2022.11.04.514231 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9
-
"Fast and accurate modeling and design of antibody-antigen complex using tFold" (tFold-Ag). bioRxiv 2024.02.05.578892, 2024. https://www.biorxiv.org/content/10.1101/2024.02.05.578892v1 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8
Comments and feedback
Spotted an error or have a counterpoint? Comment below. No account needed, a name is enough. Corrections and pushback are welcome.