How AlphaGenome Tackles Variant Effect Prediction
The human genome has 3 billion base pairs. Less than 2% of them code for proteins 1. The remaining 98% contains regulatory instructions that control when and where genes turn on 2. Most disease-associated variants identified by genome-wide association studies sit in these non-coding regions, affecting gene expression rather than protein sequence 3. The problem: given a single-nucleotide change somewhere in those 3 billion letters, predict what breaks.
This is variant effect prediction (VEP). Each person carries roughly 4 to 5 million single nucleotide polymorphisms 4. Scientists have catalogued over 600 million globally 5. Most have no effect on health 6, but the ones that matter can alter regulatory logic in ways no short-context model can detect.
AlphaGenome, published in Nature in January 2026 by Google DeepMind, changes the calculus. It processes up to 1 million DNA base pairs and outputs predictions about thousands of molecular properties related to gene regulation 7. It scored state-of-the-art on 22 of 24 DNA sequence prediction tasks and matched or exceeded top-performing models on 25 of 26 variant-effect evaluations 8. Variant scoring takes roughly one second 9.
 Source: Google DeepMind, "AlphaGenome: AI for better understanding the genome"
Source: Google DeepMind, "AlphaGenome: AI for better understanding the genome"
Why Context Length Matters
Gene regulation operates over enormous distances. Enhancers can activate gene expression from thousands or even millions of base pairs away 10. If your model only sees a small window, it will miss these interactions entirely.
SpliceAI, one of the strongest specialized tools, evaluates 10,000 nucleotides of flanking sequence context 11 and achieves 95% accuracy predicting splice junctions 12. Excellent for local splicing, but blind to long-range regulation. Enformer pushed context to 200,000 base pairs, five times Basenji2's 40,000 base-pair capability 1314. It was significantly more accurate at predicting variant effects on gene expression 15, but 200kb still covers only a fraction of known regulatory distances.
 SpliceAI's 10k context window captures local splice signals but misses long-range regulatory interactions. Source: Illumina / Jaganathan et al., Cell 2019
SpliceAI's 10k context window captures local splice signals but misses long-range regulatory interactions. Source: Illumina / Jaganathan et al., Cell 2019
Foundation models pushed context windows further. HyenaDNA reached 1 million tokens at single nucleotide resolution 16, training 160x faster than Transformer-based approaches 17. DNABERT-2 achieved comparable performance with 21x fewer parameters and approximately 92x less GPU time 18. But a benchmarking study found that current genomic language models do not offer substantial advantages over conventional machine learning approaches that use one-hot encoded sequences 19. Task-specific supervised training may still be necessary for high-performance regulatory genomics prediction 20.
AlphaGenome takes the supervised route. Rather than pre-train and hope the model captures regulatory grammar, it trains directly on experimental data from ENCODE 21 and predicts across all major regulatory modalities in a single pass.
Architecture: Three Components at Scale
AlphaGenome's design follows from two requirements: "long sequence context is important for covering regions regulating genes from far away and base-resolution is important for capturing fine-grained biological details" 22. The architecture combines:
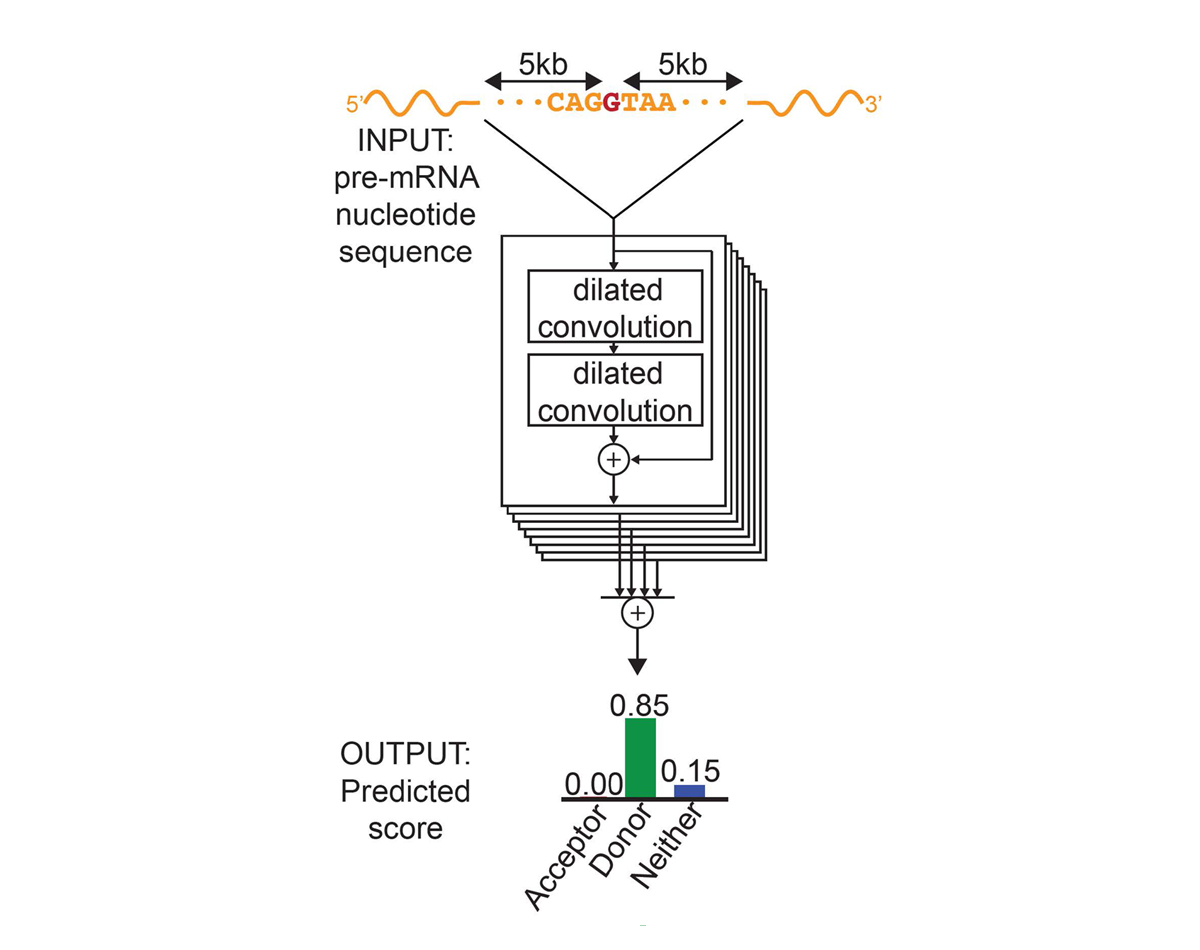
- Convolutional layers for detecting short genomic patterns like transcription factor binding motifs and splice signals 23
- Transformer layers for communicating information across sequence positions, capturing long-range enhancer-promoter interactions 24
- Distributed computation across multiple TPUs to make the 1M base-pair input tractable 25
Training required four hours and consumed half the computational resources used for the earlier Enformer model 26. More context, more output modalities, less compute.
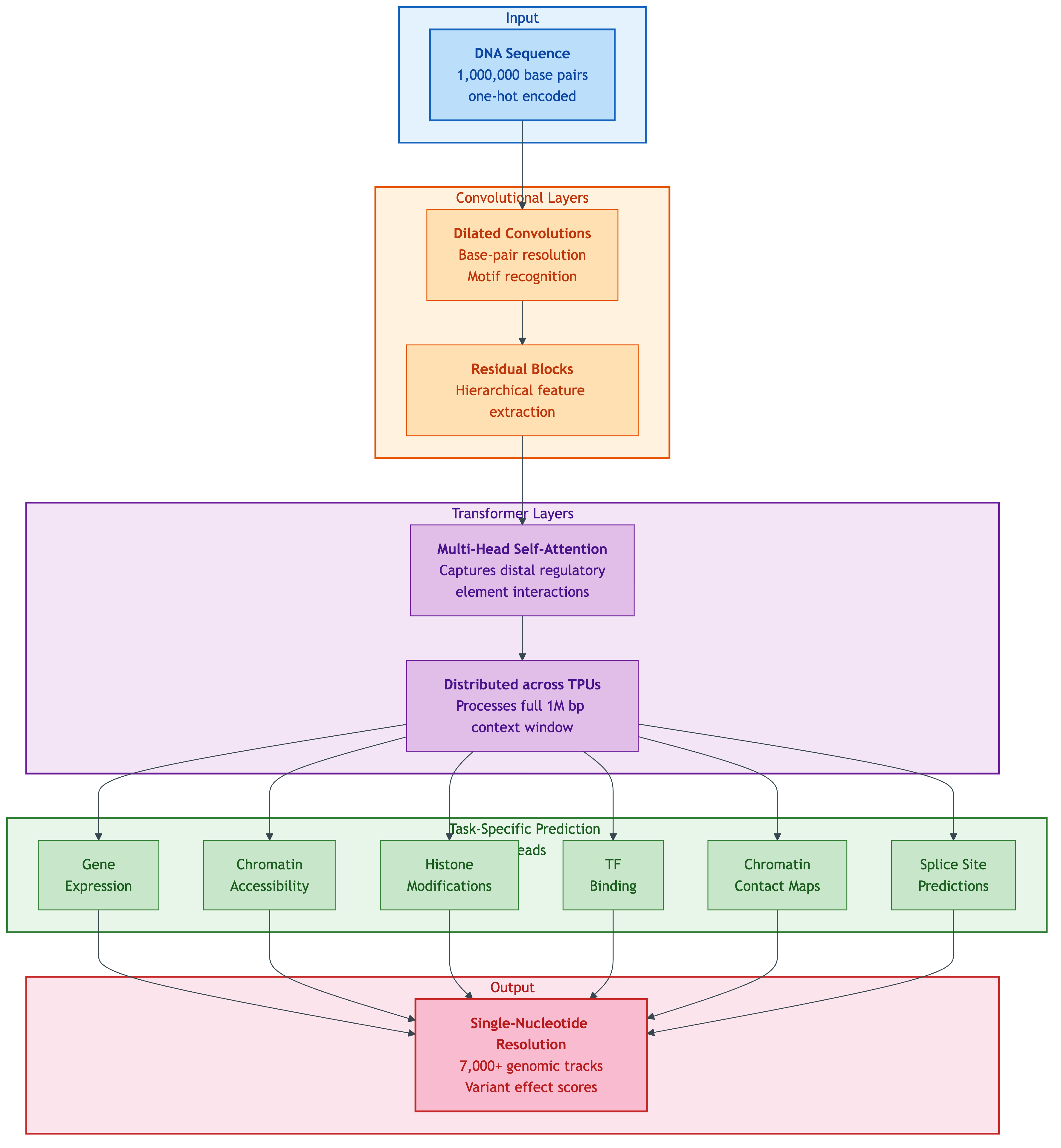 AlphaGenome processes 1M base pairs through convolutional layers (local pattern detection), transformer layers (long-range context via multi-head self-attention across TPUs), and six task-specific prediction heads to produce single-nucleotide resolution output across 7,000+ genomic tracks. Source: Google DeepMind, AlphaGenome blog
AlphaGenome processes 1M base pairs through convolutional layers (local pattern detection), transformer layers (long-range context via multi-head self-attention across TPUs), and six task-specific prediction heads to produce single-nucleotide resolution output across 7,000+ genomic tracks. Source: Google DeepMind, AlphaGenome blog
For the first time, the model explicitly predicts RNA splice-junction locations and expression levels directly from DNA sequence 27. This matters because cryptic splice variants comprise approximately 9-11% of pathogenic mutations in rare genetic disorders 28, and previous tools either ignored splicing or handled it with a separate model.
Variant Scoring: Feed In, Change, Subtract
The scoring workflow is simple in concept. Feed the reference sequence through the model. Feed the same sequence with one nucleotide changed. Subtract the output vectors. The difference across all 7,000+ tracks is the variant effect profile. The system evaluates a variant's impact across all predicted properties within approximately one second 9.
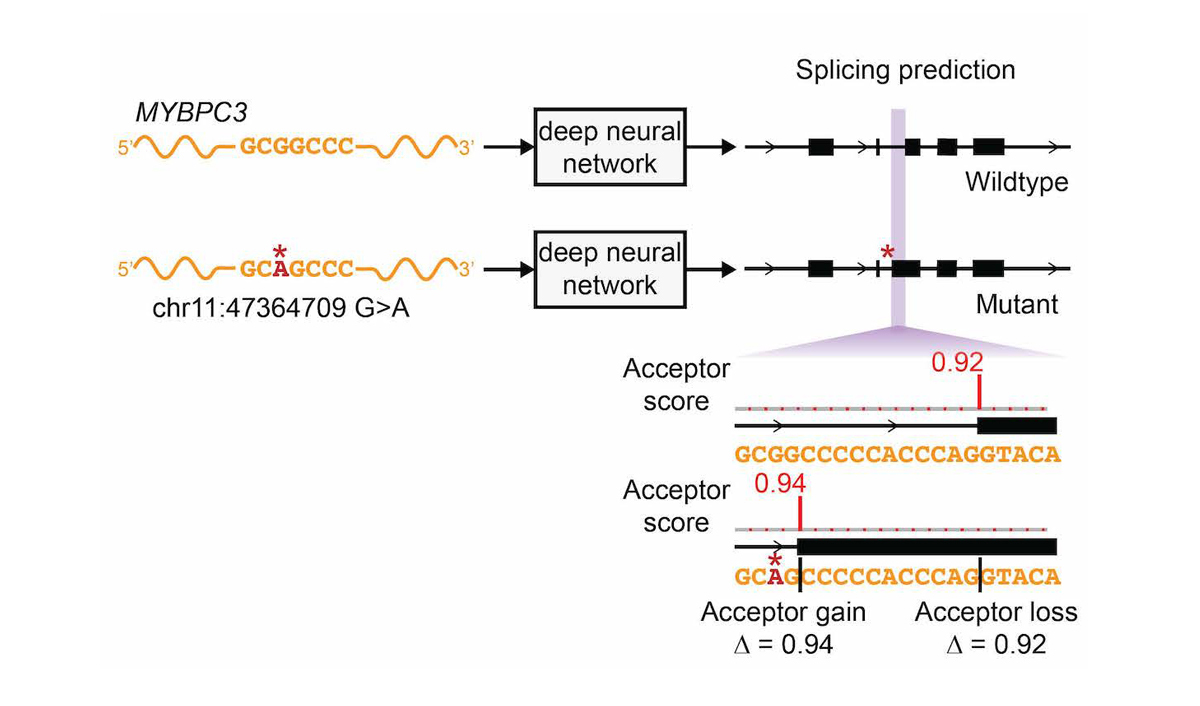 Delta-score calculation illustrated with SpliceAI's assessment of a pathogenic MYBPC3 variant. AlphaGenome extends this principle across 7,000+ prediction tracks simultaneously. Source: Illumina / Jaganathan et al., Cell 2019
Delta-score calculation illustrated with SpliceAI's assessment of a pathogenic MYBPC3 variant. AlphaGenome extends this principle across 7,000+ prediction tracks simultaneously. Source: Illumina / Jaganathan et al., Cell 2019
from alphagenome.data import genome
from alphagenome.models import dna_client
import numpy as np
model = dna_client.create("YOUR_API_KEY")
interval = genome.Interval(chromosome="chr1", start=47_183_000, end=48_183_000)
variant = genome.Variant(
chromosome="chr1", position=47_683_800,
reference_bases="T", alternate_bases="C", # TAL1 regulatory SNP
)
outputs = model.predict_variant(interval=interval, variant=variant)
ref_tracks = outputs.reference.tracks # shape: (seq_len, num_tracks)
alt_tracks = outputs.alternate.tracks
delta = np.mean(np.abs(ref_tracks - alt_tracks), axis=0)
top5_idx = np.argsort(delta)[::-1][:5]
for rank, idx in enumerate(top5_idx, 1):
print(f" {rank}. {outputs.track_metadata.names[idx]:.<50s} delta={delta[idx]:.4f}")
The model demonstrated clinical relevance by predicting that certain cancer-associated mutations would activate the TAL1 gene by introducing a MYB DNA binding motif, successfully replicating the known disease mechanism 29. Not just a pathogenicity score, but an explanation of how the variant acts.
How AlphaGenome Compares
The comparison set includes specialized models that each excel at one modality. Here is how the field stacks up:
| Model | Context | Key Capability | Trade-off |
|---|---|---|---|
| SpliceAI | 10k nt | 95% splice junction accuracy 12 | Splicing only, no long-range |
| Enformer | 200k bp | Expression + chromatin VEP 15 | No splicing, limited context |
| HyenaDNA | 1M tokens | Sub-quadratic scaling 30 | Foundation model, not VEP-specific |
| DNABERT-2 | Variable | 21x fewer params, 92x less GPU 18 | General tasks, short context |
| AlphaMissense | Protein-level | 89% of 71M missense variants classified 31 | Coding regions only (2% of genome) |
| AlphaGenome | 1M bp | 7,000+ tracks, all modalities, ~1s/variant 79 | TPU-dependent, API-only access |
AlphaGenome's key differentiator: it is the only model that jointly predicts all assessed regulatory modalities. AlphaMissense covers the 2% of the genome encoding proteins. AlphaGenome covers the other 98%, the non-coding regulatory regions containing numerous disease-linked variants 32.
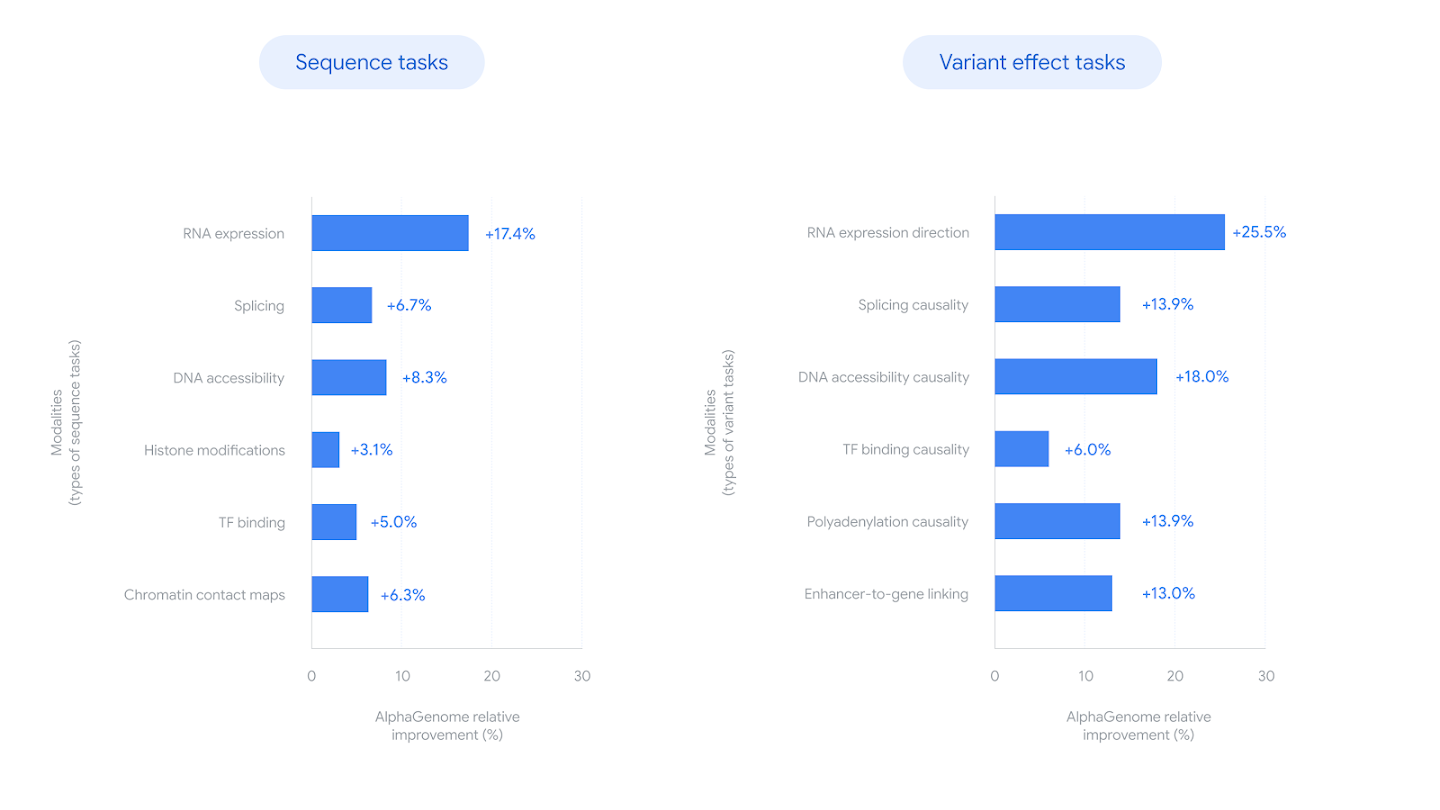 AlphaGenome's relative improvements on DNA sequence and variant effect prediction tasks compared to existing state-of-the-art methods. Source: Google DeepMind
AlphaGenome's relative improvements on DNA sequence and variant effect prediction tasks compared to existing state-of-the-art methods. Source: Google DeepMind
Limitations
AlphaGenome is honest about its boundaries. Capturing very distant regulatory elements, beyond 100,000 DNA letters away, remains challenging 33. The 1M base-pair context window is large but not infinite. Some enhancers operate across megabase distances.
The API throughput caps matter too. Query rates fluctuate based on demand, making it suitable for medium-scale analyses requiring up to thousands of predictions rather than million-scale studies 34. Scoring all 4-5 million SNPs in one genome is not yet practical through the API.
The access model also limits flexibility. The software uses Apache 2.0 licensing and documentation employs CC-BY 4.0 35, but the model runs on DeepMind infrastructure. Researchers who need to run ablations, fine-tune for new cell types, or process millions of variants face constraints that open-weight models like HyenaDNA or DNABERT-2 do not impose.
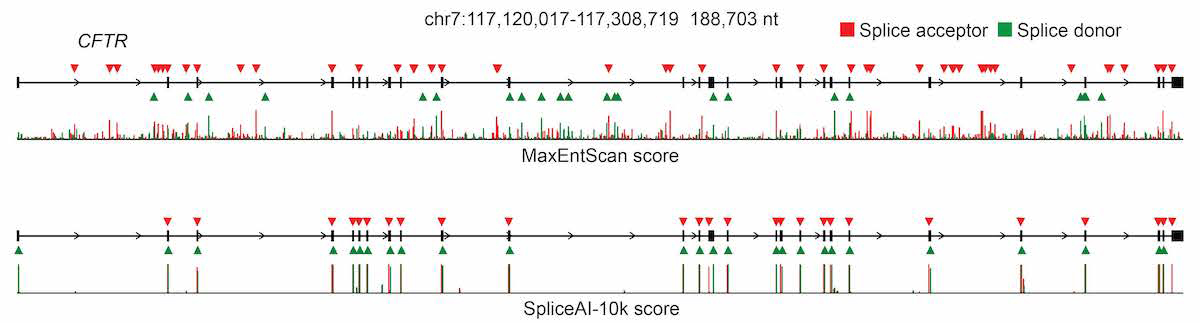 Traditional vs. deep learning splice prediction for the CFTR gene. SpliceAI (bottom) provides far more accurate exon boundary prediction than MaxEntScan (top), illustrating the trajectory AlphaGenome extends further. Source: Illumina / Jaganathan et al., Cell 2019
Traditional vs. deep learning splice prediction for the CFTR gene. SpliceAI (bottom) provides far more accurate exon boundary prediction than MaxEntScan (top), illustrating the trajectory AlphaGenome extends further. Source: Illumina / Jaganathan et al., Cell 2019
What This Means
For the first time, a single model covers long-range context (1M bp), multi-modal output (7,000+ tracks), and fast inference (~1 second per variant). A clinical geneticist can take a non-coding variant of uncertain significance, score it against splicing, expression, chromatin, and transcription factor binding predictions in one query, and get a mechanistic hypothesis for how the variant might cause disease.
The tool helps scientists understand how single-letter mutations and distant DNA regions influence gene activity, shaping health and disease risk 36. The limitations are real: the >100k blind spot, API throughput caps, and the lack of local deployment all constrain adoption. But the 98% of the genome that does not encode proteins is no longer invisible to deep learning.
AlphaGenome is one model, not the last model.
References
Footnotes
-
Avsec et al., Google DeepMind Enformer blog. ↩
-
Avsec et al., Google DeepMind Enformer blog. ↩
-
NHGRI, Gene Expression and Regulation. ↩
-
NHGRI, Single Nucleotide Polymorphisms. ↩
-
NHGRI, Single Nucleotide Polymorphisms. ↩
-
NHGRI, Single Nucleotide Polymorphisms. ↩
-
Google DeepMind, AlphaGenome blog post, January 2026. https://deepmind.google/blog/alphagenome-ai-for-better-understanding-the-genome/ ↩ ↩2
-
Google DeepMind / Avsec et al., Nature (2026). DOI: 10.1038/s41586-025-10014-0 ↩
-
NHGRI, Gene Expression and Regulation. ↩
-
Jaganathan et al., Cell (2019). ↩
-
Avsec et al., Nature Methods (2021). ↩
-
Avsec et al., Nature Methods (2021). ↩
-
Nguyen et al., "HyenaDNA," NeurIPS (2023). ↩
-
Nguyen et al., "HyenaDNA," NeurIPS (2023). ↩
-
Tang, Somia, Yu, and Koo, bioRxiv (2024). DOI: 10.1101/2024.02.29.582810 ↩
-
Tang, Somia, Yu, and Koo, bioRxiv (2024). ↩
-
ENCODE Project. ↩
-
Google DeepMind, AlphaGenome blog. ↩
-
Google DeepMind, AlphaGenome blog. ↩
-
Google DeepMind, AlphaGenome blog. ↩
-
Google DeepMind, AlphaGenome blog. ↩
-
Google DeepMind, AlphaGenome blog. ↩
-
Google DeepMind, AlphaGenome blog. ↩
-
Jaganathan et al., Cell (2019). ↩
-
Google DeepMind, AlphaGenome blog. ↩
-
Nguyen et al., "HyenaDNA," NeurIPS (2023). ↩
-
Cheng et al., "AlphaMissense," Science (2023). ↩
-
Google DeepMind, AlphaGenome blog. ↩
-
Google DeepMind, AlphaGenome blog. ↩
-
Google DeepMind, AlphaGenome GitHub. https://github.com/google-deepmind/alphagenome ↩
-
Google DeepMind, AlphaGenome GitHub. ↩
-
Saey, T.H., Science News, January 28, 2026. ↩