The pitch for genomic foundation models is that one pretrained network now beats task-specific tools across the board, from regulatory annotation to clinical variant interpretation. I have spent the last six months reading the 2025-2026 literature with that claim in mind, and the honest answer is that it is true for exactly one family of tasks and false for several others that get the same marketing. For variant effect prediction, the frontier models are genuinely closing the gap with specialist tools, and on some noncoding benchmarks they have passed them: Evo 2 set a new state of the art for BRCA1 noncoding single-nucleotide variants without any task-specific training,1 and AlphaGenome matched or exceeded the strongest available external model on 24 of 26 variant-effect evaluations.2 For perturbation prediction and for mechanistic interpretation of attention, the strongest published evidence is that deliberately trivial baselines still win: five foundation models plus two other deep networks failed to beat simple linear baselines on perturbation response, and none beat them in any setting tested.3 Those two facts live in the same field, often in the same model.
It helps to keep two ledgers while reading this literature. A capability ledger records what the models can demonstrably do at scale, and a validity ledger records what holds up when you pass each claim through an independent test set with an honest baseline. The marketing reports the first ledger; a molecular pathology lab has to act on the second. So the organizing question here is not "which model is best." It is: when you subtract leaderboard theatre and force every claim through a held-out test set with a baseline the model has to beat, what is left standing?
This is an update to a guide I wrote in January 2026 that compared DNABERT-2, HyenaDNA, Nucleotide Transformer, ESM-2, ESMFold, scGPT, Geneformer, and the original Evo. The field has moved since then. Evo 2 (40 billion parameters, a 1-million-token single-nucleotide context) and AlphaGenome (1 megabase of input at base-pair resolution) arrived; Caduceus made a serious case that architecture beats scale; ESM3 and RoseTTAFold All-Atom pushed structure models toward multimodal, all-atom biomolecular design; and a substantial skeptical literature, including a unified benchmark over 40 genomic models and a Nature Methods negative result, gave us the tools to separate measured capability from leaderboard theatre. This piece is organized around the evidence, not the vendors. The goal is the comparison a molecular pathologist can forward to a colleague because it is fair to each model and explicit about what is not yet known.
Why an honest comparison is hard, before any results
The single most important 2026 result for this audience is not a model. It is GENEB, a diagnostic benchmark that evaluated frozen representations from 40 genomic foundation models across 100 tasks in 13 functional categories under one unified probing protocol.4 Its conclusion is uncomfortable and worth stating before any per-model number: aggregate leaderboards are unstable, model rankings vary sharply across task categories, scale provides only modest and inconsistent gains, and architectural and pretraining alignment frequently outweigh parameter count.4 The same model is sometimes characterized as a breakthrough in one paper and an underperformer in another, reflecting not contradictory evidence but the absence of a common evaluation framework.4 The reason that finding is hard to wave away is that none of the 40 models GENEB evaluated was built by its authors or their funders.4 There is no vendor incentive in the result.
If you do not address the field's known traps up front, an expert reader assumes you do not know about them. There are five that matter for clinical genomics.
Tokenization is not neutral. DNA models disagree on what a token even is. HyenaDNA and the Evo family operate at single-nucleotide, byte-level resolution.56 DNABERT-2 replaced k-mer tokenization with Byte Pair Encoding, which iteratively merges the most frequent co-occurring genome segments into a learned vocabulary.7 The DNABERT-2 authors made this change in part because overlapping k-mer schemes leak information across adjacent tokens, an artifact that can inflate a downstream score without any real learning.7 When two models report the same accuracy on the same task, they may be solving slightly different problems at slightly different granularity, and the resolution at which a model tokenizes constrains the resolution at which it can call a variant.
 Figure 1: Illustration of k-mer tokenization drawbacks (information leakage in the overlapping setting) that motivated Byte Pair Encoding in DNABERT-2. Source: DNABERT-2 (Zhou et al., ICLR 2024)
Figure 1: Illustration of k-mer tokenization drawbacks (information leakage in the overlapping setting) that motivated Byte Pair Encoding in DNABERT-2. Source: DNABERT-2 (Zhou et al., ICLR 2024)
Context length is asymmetric and consequential. Early Transformer DNA models were limited to 512 to 4,000 tokens, less than 0.001 percent of the human genome, which structurally prevented them from modeling long-range interactions.5 That matters clinically because nucleic acids up to one million base pairs from a gene can have significant regulatory effects.8 A model with a 4 kb window and a model with a 1 Mb window are not competing on a level field for an enhancer-promoter question. Comparing them without saying so is misleading, and many published comparisons do exactly that.
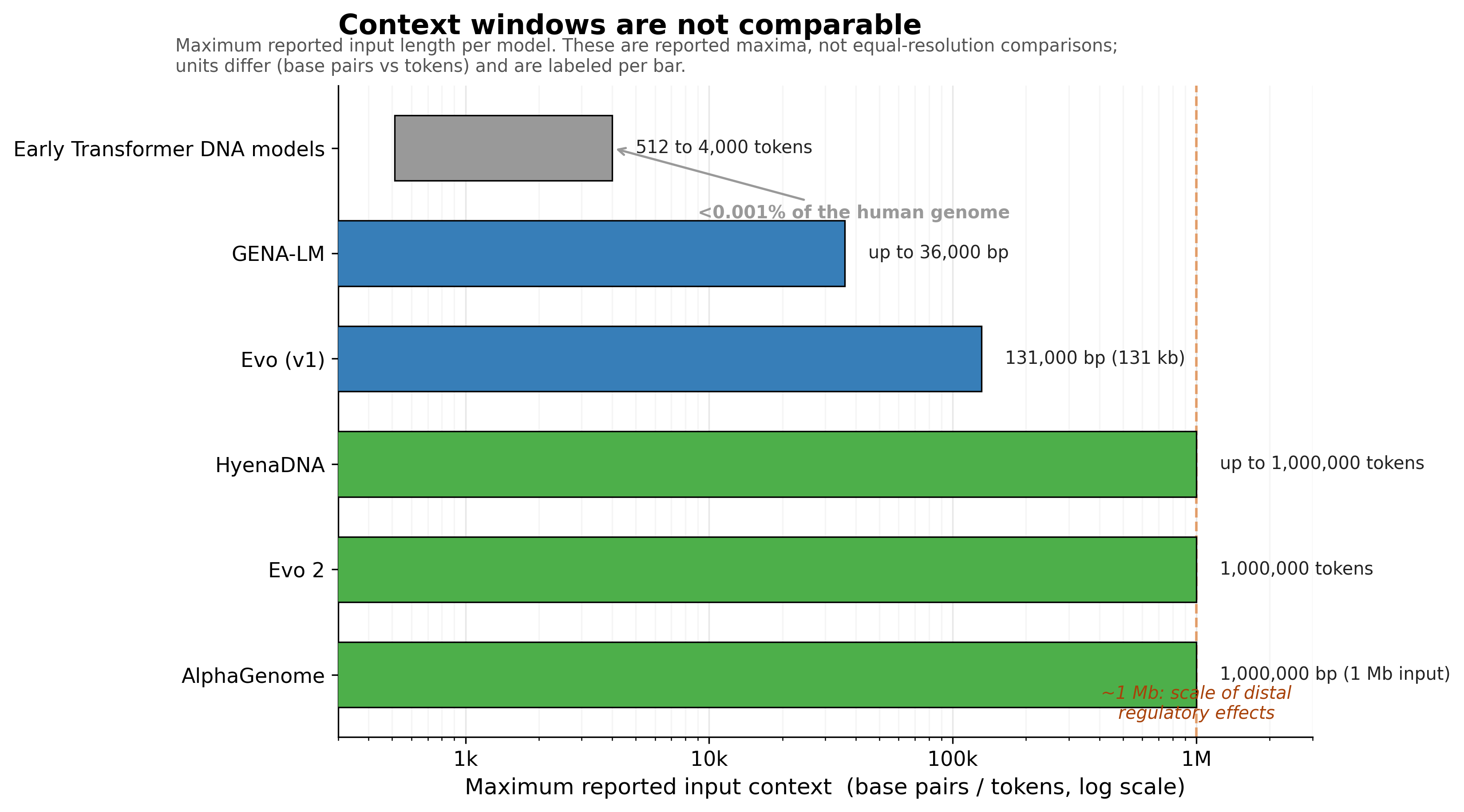 Figure 2: Reported maximum context windows across genomic models, log scale. These are reported maxima, not equal-resolution comparisons, and units differ across models (tokens versus base pairs). Sources: HyenaDNA5, Evo6, Evo 21, AlphaGenome2, GENA-LM4.
Figure 2: Reported maximum context windows across genomic models, log scale. These are reported maxima, not equal-resolution comparisons, and units differ across models (tokens versus base pairs). Sources: HyenaDNA5, Evo6, Evo 21, AlphaGenome2, GENA-LM4.
Contamination against public references is the default, not the exception. These models are pretrained on GRCh38 and large public collections, and they are then evaluated against benchmarks built from the same references, ClinVar, and gnomAD. Retrospective AUROC on a ClinVar split tells you how well a model recapitulates labels that may already be encoded in its training distribution. It does not tell you how the model behaves on a genuinely novel variant in a real case. The single-cell literature has shown the mechanism concretely: scGPT and Geneformer do not consistently outperform baselines even on datasets they saw during pretraining.9 This gap, between retrospective discrimination and prospective clinical utility, is the single most important caveat in the whole field.
Benchmarks are fragmented and unstable. The DNABERT-2 authors identified the absence of a standardized benchmark as a significant impediment to fair comparison and built the GUE suite (36 datasets, 9 tasks, input lengths from 70 to 10,000 bp) in response.7 BEND made the same diagnosis for long-range tasks: evaluation tasks differ between works and often fail to recapitulate the length, scale, and sparsity of real genome annotation.10 OmniGenBench reaches the same conclusion about inconsistent metrics across studies.11 GENEB's task-group heatmap is the clearest single picture of the problem: rankings reshuffle as you move across functional categories, with no model dominant everywhere.
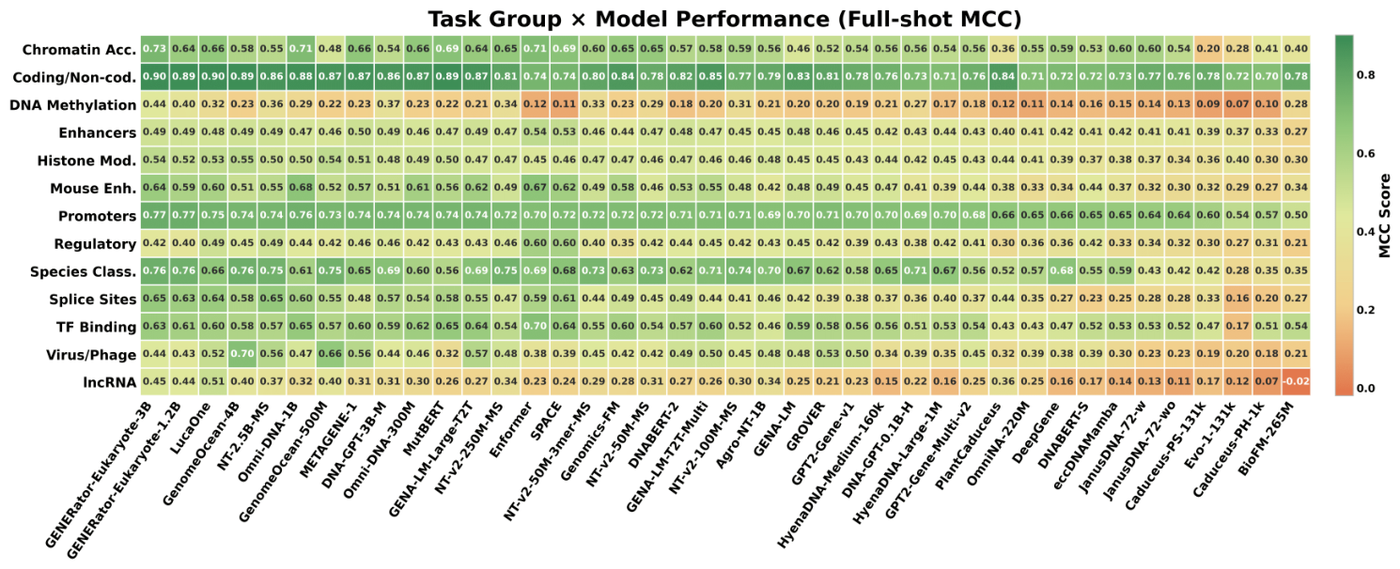 Figure 3: Full-shot Matthews correlation coefficient across 40 genomic foundation models and 13 task groups; the leaderboard is category-dependent, not absolute. Source: GENEB (Ledneva et al., arXiv:2606.04525, 2026)
Figure 3: Full-shot Matthews correlation coefficient across 40 genomic foundation models and 13 task groups; the leaderboard is category-dependent, not absolute. Source: GENEB (Ledneva et al., arXiv:2606.04525, 2026)
DNA is not protein, and the benchmark history shows it. Protein structure prediction had CASP, a curated competition the field credits with making AlphaFold's progress legible.12 Genomics has historically lacked an equivalent, with similar challenges (genome annotation, functional element identification) but no folding-competition analogue.12 Part of why DNA models lag protein models on benchmarking maturity is biological: in DNA the signal can span an extremely long range, high-signal regions are sparse, and even within them the signal density is lower than in proteins.10 BEND's own finding is that current DNA language model embeddings can approach expert methods on some tasks but capture only limited information about long-range features.10 Keep that asymmetry in mind whenever a DNA result is presented with protein-level confidence.
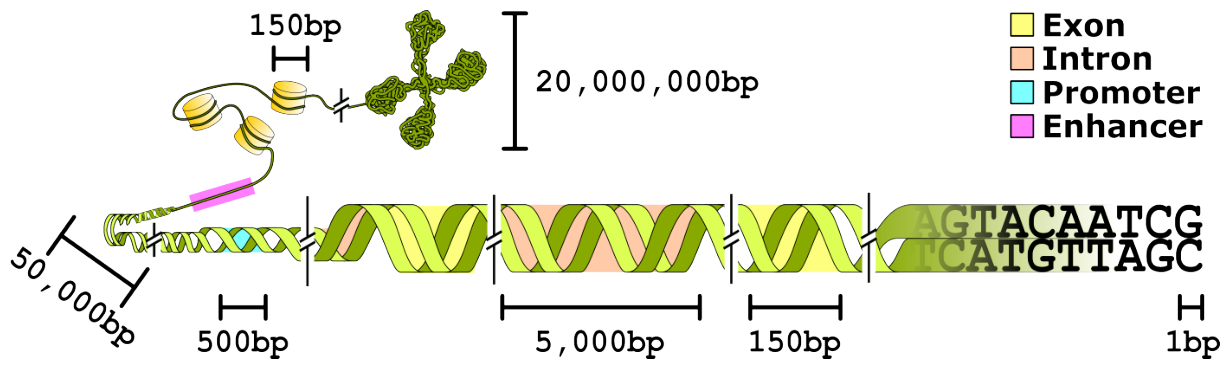 Figure 4: Organization of eukaryotic genomic DNA with indicative length scales, used to frame BEND's genome-annotation tasks and the long-range-feature challenge. Source: BEND (Marin et al., ICLR 2024)
Figure 4: Organization of eukaryotic genomic DNA with indicative length scales, used to frame BEND's genome-annotation tasks and the long-range-feature challenge. Source: BEND (Marin et al., ICLR 2024)
With those caveats established, here is the map.
A map of the field
It helps to see the field as an architectural family tree before drilling into individual models. GENEB's taxonomy is a useful orientation: genomic foundation models split into Transformer-based families, state-space (Mamba) families, and convolution-hybrid families, and that architectural lineage turns out to predict task fit better than parameter count does.4
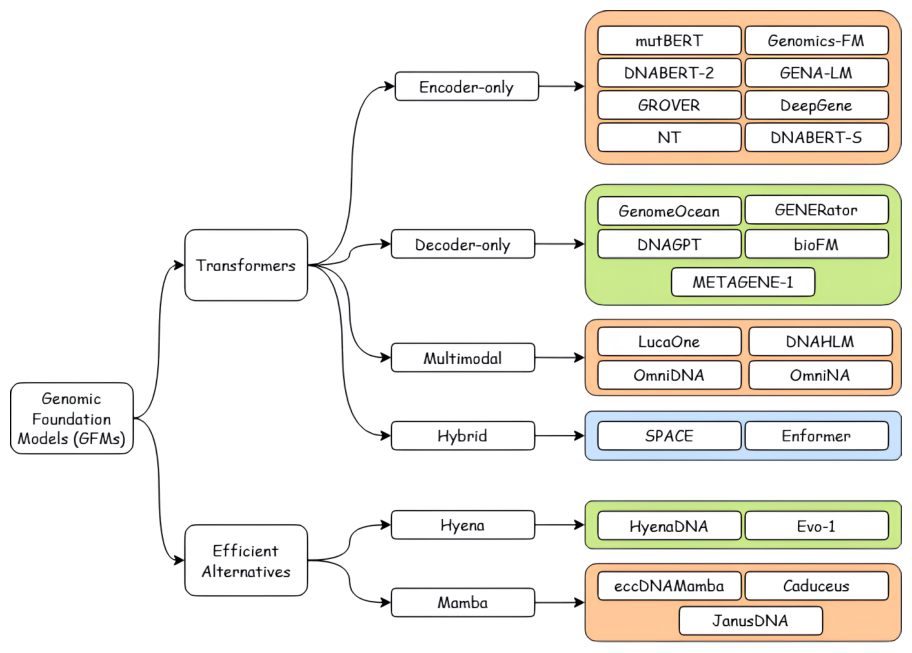 Figure 5: Architectural taxonomy of genomic foundation models, a map of the field before the per-model analysis. Source: GENEB (Ledneva et al., arXiv:2606.04525, 2026)
Figure 5: Architectural taxonomy of genomic foundation models, a map of the field before the per-model analysis. Source: GENEB (Ledneva et al., arXiv:2606.04525, 2026)
Two design axes dominate the DNA side. The first is scale plus long context: bigger models with longer windows, exemplified by Evo 2 and AlphaGenome. The second is efficient, biologically principled architecture: smaller models whose inductive biases match the structure of DNA, exemplified by Caduceus and HyenaDNA. The central empirical question for a clinical audience is which axis actually delivers, and GENEB's answer is that architectural and pretraining alignment frequently outweigh parameter count.4 That is the through-line for everything below. A third axis matters more for the clinic than either: access. Open weights you can freeze and audit are a different proposition from an API behind a vendor's terms, and that distinction is not a footnote for a regulated lab.
The capability ledger: DNA and genome-scale models
Evo 2: the scale-and-context frontier
Evo 2 is the largest DNA foundation model to date, trained on 9.3 trillion DNA base pairs from a curated genomic atlas spanning all domains of life.1 It comes in 7B and 40B parameter versions with a 1-million-token context window at single-nucleotide resolution.1 The 7B model was trained on 2.4 trillion tokens and the 40B on 9.3 trillion.1 Training used a two-phase schedule: an 8,192-token context focused on genic windows, followed by midtraining that extended the context to 1 million tokens.1 The architecture is StripedHyena 2, a hybrid of convolution and attention operators that, at 40B parameters, delivers up to a 1.3x throughput speedup at 16k context and a 3x speedup at 1 million context relative to optimized Transformer baselines.1 On a synthetic needle-in-a-haystack test, Evo 2 can retrieve a 100 bp needle from arbitrary positions inside a 1-million-bp haystack of random DNA.1 It is also one of the largest fully open AI models in any domain, with weights, code, and the OpenGenome2 dataset (over 8.8 trillion nucleotides) released.1
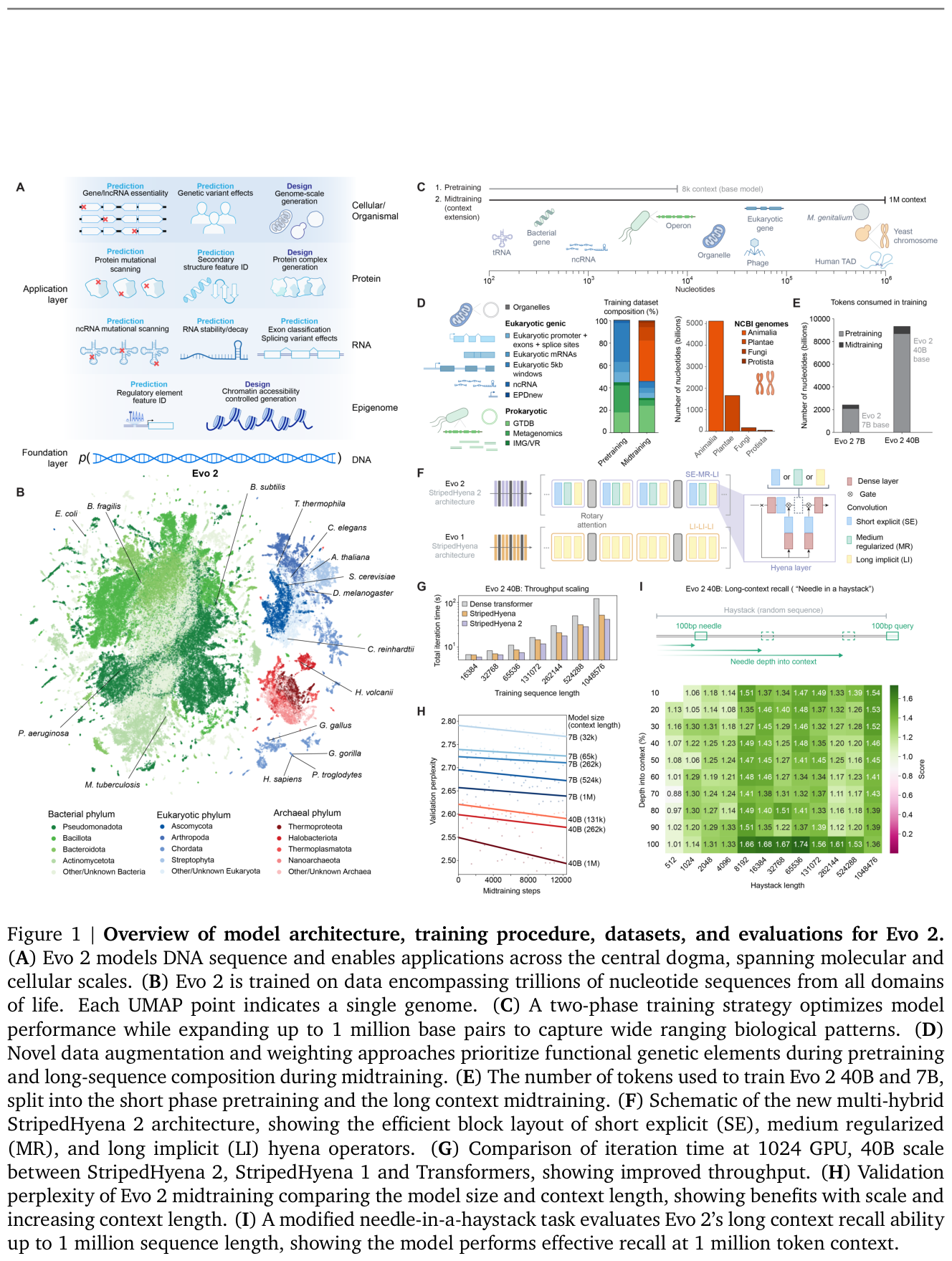 Figure 6: Evo 2 architecture, training data, and evaluation overview (40B parameters, 1M-token single-nucleotide context). Source: Evo 2 (Brixi et al., bioRxiv 2025 / Nature 2026)
Figure 6: Evo 2 architecture, training data, and evaluation overview (40B parameters, 1M-token single-nucleotide context). Source: Evo 2 (Brixi et al., bioRxiv 2025 / Nature 2026)
What it can do clinically is the interesting part, and it is also where Evo 2's authors are commendably honest. Evo 2 learns from DNA sequence alone to predict functional impacts of genetic variation without task-specific finetuning.1 But finetuning-free does not mean uniformly best. For single-nucleotide variants in coding regions, the 40B and 7B models ranked fourth and fifth, behind AlphaMissense, ESM-1b, and GPN-MSA.1 For noncoding variants, Evo 2 surpassed other models on both SNVs and non-SNVs,1 achieved the highest zero-shot performance on exonic and intronic splice variant effect prediction,1 and set a new state of the art for BRCA1 noncoding SNVs.1 An embedding-based exon classifier reached AUROCs of 0.82 to 0.99 across species.1 One negative result deserves repeating because it is rarely volunteered: Evo 2 showed no correlation between its likelihood and viral protein fitness for viruses that infect human hosts, a consequence of deliberately excluding those sequences from training.1 That is the honesty profile this audience should reward.
AlphaGenome: regulatory variant effect at base-pair resolution
AlphaGenome takes a different position on the scale-and-context axis. It ingests 1 megabase of DNA and predicts thousands of functional genomic tracks up to single-base-pair resolution across diverse modalities.2 Trained on human and mouse genomes, it simultaneously predicts 5,930 human or 1,128 mouse genome tracks across 11 modalities covering gene expression, splicing, chromatin state, and chromatin contact maps.2 On variant effect prediction it matched or exceeded the strongest available external models on 24 of 26 evaluations, and reached state of the art on 22 of 24 genome-track prediction tasks.2 One caution on that headline: 24 of 26 is a count of evaluations won, not an effect size, so it tells you how often AlphaGenome led, not by how much.
 Figure 7: AlphaGenome architecture and evaluation overview (1 Mb input, base-pair resolution, multimodal output tracks). Source: AlphaGenome (Avsec et al., bioRxiv 2025 / Nature 2026)
Figure 7: AlphaGenome architecture and evaluation overview (1 Mb input, base-pair resolution, multimodal output tracks). Source: AlphaGenome (Avsec et al., bioRxiv 2025 / Nature 2026)
AlphaGenome is built to resolve a real tradeoff in the prior literature. Base-resolution models like SpliceAI were restricted to short inputs (10 kb or less), missing distal regulatory elements; longer-context models like Enformer and Borzoi reached 200 kb to 500 kb but only at reduced 128 bp or 32 bp output resolution.2 AlphaGenome's claim is to get both at once. It does so through a two-stage training process of pre-training then distillation, where a single student model is trained to reproduce an ensemble of teachers, and that student runs in under one second per variant on an NVIDIA H100.2 Base-pair training over the full 1 Mb sequence required sequence parallelism across 8 interconnected TPUv3 devices.2 The clinical relevance is direct: over 98 percent of observed human genetic variation is noncoding, and characterizing it globally is intractable without computational prediction.2 AlphaGenome is strongest exactly there, including splicing.
Two caveats belong on the same page as the headline. The AlphaGenome authors themselves note that generalist models can lag specialists on certain tasks, like splicing, or lack particular modalities, such as contact maps.2 And AlphaGenome is API-only and non-commercial. For a diagnostic lab that needs to validate, version-lock, and reproduce a pipeline under a quality system, an API behind a vendor's terms is a materially different proposition from open weights you can freeze and audit. That is not a knock on the science. It is a deployment constraint that should weigh heavily in clinical settings.
Caduceus: the case that architecture beats scale
If Evo 2 and AlphaGenome are the scale argument, Caduceus is the counterargument, and it is the one I find most instructive. Caduceus is the first family of reverse-complement (RC) equivariant, bi-directional, long-range DNA language models, built on Mamba state-space modules rather than attention.8 The biological motivation is clean: DNA has two strands that are reverse complements carrying the same information, and building that symmetry into the model as an inductive bias improves performance.8 Mamba blocks handle sequences of hundreds of thousands of nucleotides without the quadratic cost of attention.8
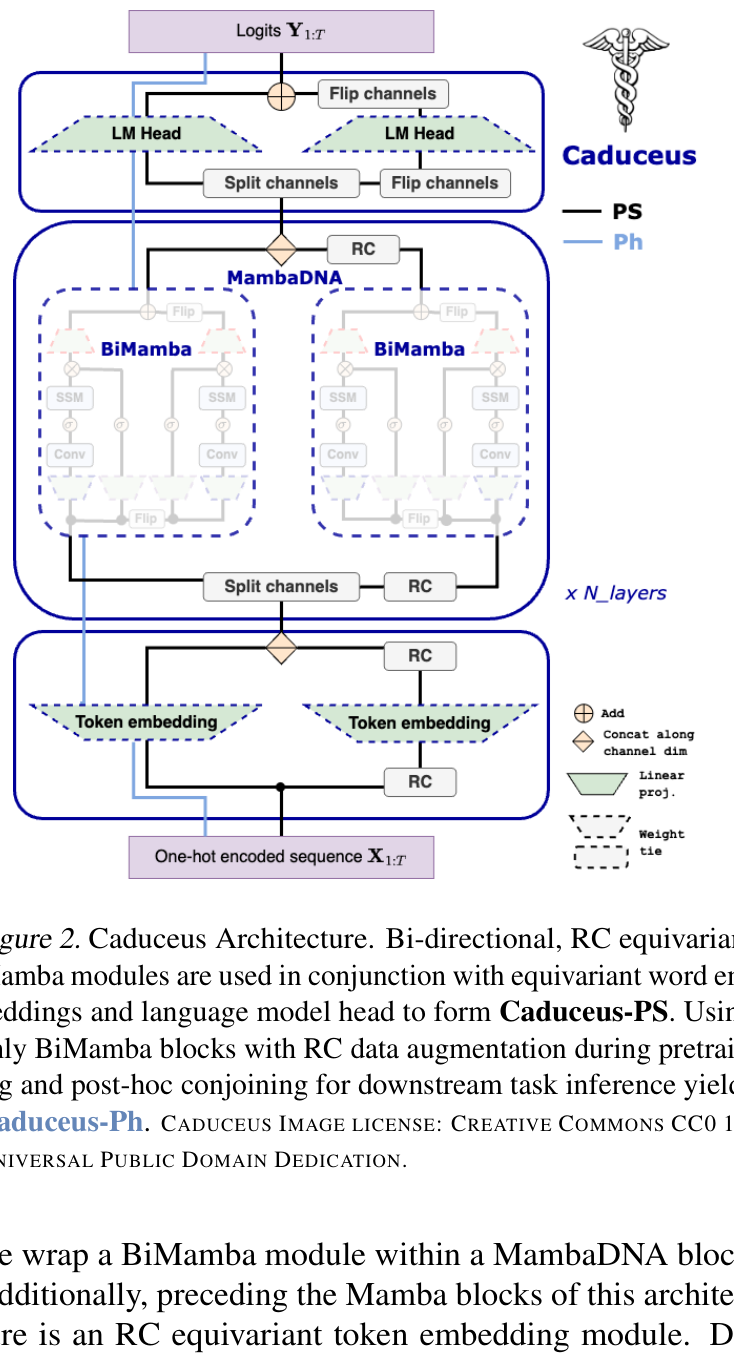 Figure 8: Caduceus architecture: bi-directional, reverse-complement equivariant stack with BiMamba layers. Source: Caduceus (Schiff et al., ICML 2024)
Figure 8: Caduceus architecture: bi-directional, reverse-complement equivariant stack with BiMamba layers. Source: Caduceus (Schiff et al., ICML 2024)
The result that should make every scaling enthusiast pause: on a challenging long-range variant effect prediction task, Caduceus exceeded the performance of models 10 times larger that did not use bi-directionality or equivariance.8 The advantage is strongest precisely where it should be, at long distances from the transcription start site.
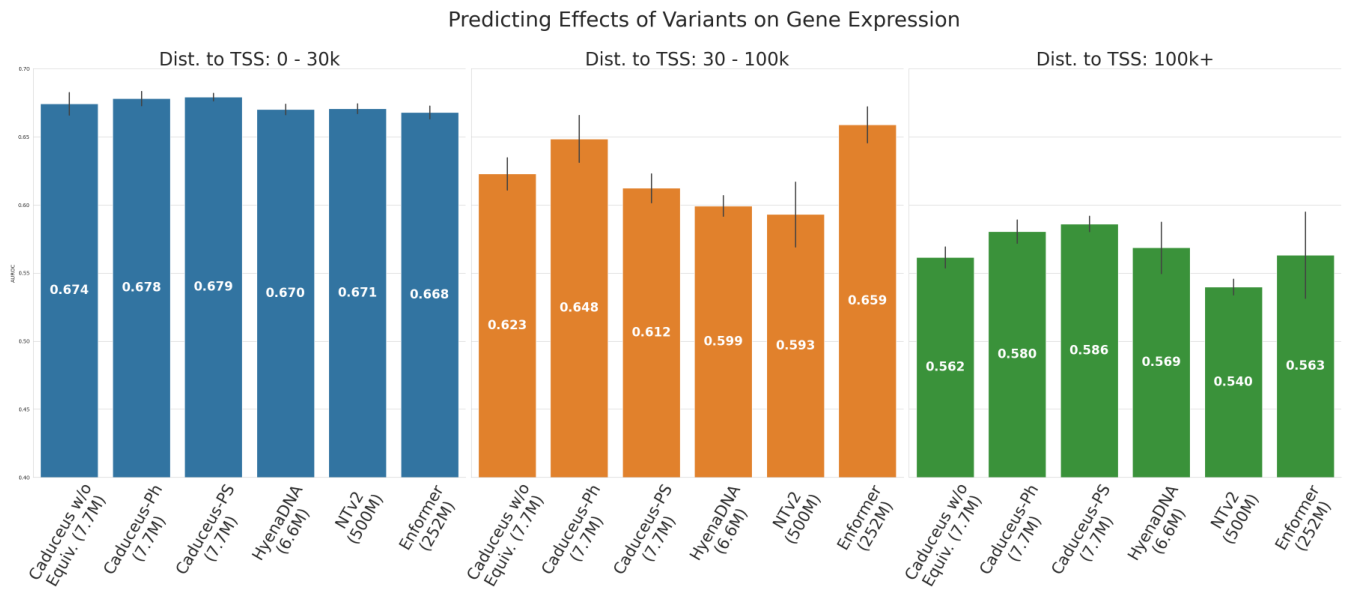 Figure 9: Variant-effect prediction on gene expression across distance bins to the nearest TSS, showing the Caduceus advantage at long range versus larger non-equivariant models. Source: Caduceus (Schiff et al., ICML 2024)
Figure 9: Variant-effect prediction on gene expression across distance bins to the nearest TSS, showing the Caduceus advantage at long range versus larger non-equivariant models. Source: Caduceus (Schiff et al., ICML 2024)
GENEB corroborates this at the level of the whole field. Its model-size-versus-performance Pareto frontier puts small, architecture-aligned models on or near the frontier while some large models sit below it.4 Parameter count is not destiny.
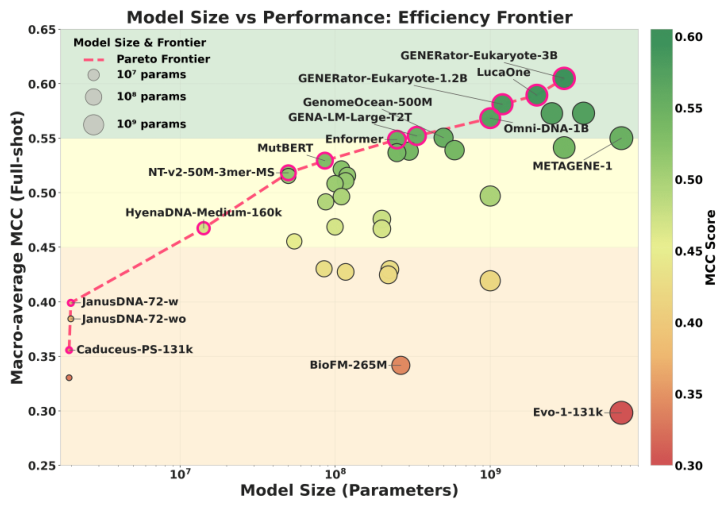 Figure 10: Model size versus macro-average MCC across 40 genomic foundation models; architecture and pretraining alignment, not parameter count, predict frontier position. Source: GENEB (Ledneva et al., arXiv:2606.04525, 2026)
Figure 10: Model size versus macro-average MCC across 40 genomic foundation models; architecture and pretraining alignment, not parameter count, predict frontier position. Source: GENEB (Ledneva et al., arXiv:2606.04525, 2026)
The efficient and open lineage: HyenaDNA, DNABERT-2, GENA-LM, Nucleotide Transformer
HyenaDNA was the proof of concept that long context need not be expensive. It reached context lengths up to 1 million tokens at single-nucleotide resolution, an up to 500x increase over previous dense attention models, while scaling sub-quadratically and training up to 160x faster than a Transformer at long sequence lengths.5 On the Nucleotide Transformer benchmark suite it reached state of the art on 12 of 18 datasets using a model with 1,500x fewer parameters than the comparison point, and 3,200x less pretraining data.5 The parameter comparison is worth stating precisely because it is often inverted: the small model here is HyenaDNA at roughly 1.6 million parameters, and the 2.5-billion-parameter baseline it beat is the Nucleotide Transformer.5 On the Genomic Benchmarks it surpassed prior state of the art on 7 of 8 datasets, by an average of 10 accuracy points and up to 20 on enhancer identification.5
DNABERT-2 made the efficiency argument on the training-cost side. With BPE tokenization it achieved performance comparable to the prior state of the art with 21x fewer parameters and roughly 92x less GPU time in pretraining,7 and it outperformed the original DNABERT on 23 of 28 GUE datasets while being 3x more efficient.7 The concrete training-budget comparison the authors report is striking in context: about 14 days on 8 NVIDIA RTX 2080Ti versus 28 days on 128 A100s for the model it was matching.7 For a lab without a hyperscaler's compute, that difference is the difference between feasible and not.
GENA-LM rounds out the open, peer-reviewed end of the spectrum. It is a suite of Transformer-based DNA models handling inputs up to 36,000 base pairs, extended further by a recurrent memory mechanism, and shipped with multispecies and taxon-specific variants that finetune with modest computational demands.13 It was published in Nucleic Acids Research in January 2025, with open weights.13 For clinical reproducibility, open and peer-reviewed is a meaningful category, and GENA-LM sits squarely in it.
The Nucleotide Transformer remains the reference point these efficiency claims are measured against, and the right way to treat it here is as the 2.5-billion-parameter baseline that smaller, better-aligned models have repeatedly matched or beaten.5 The framing has shifted since 2024: it is no longer obvious that a larger Transformer trained on more genomes is the right default, and GENEB's instability finding is in large part a finding about models built on that assumption.4
Variant effect prediction: where the two ledgers nearly agree
If there is one task where foundation models have earned a place in the clinical conversation, it is variant effect prediction, and the reason is that this is the task with a real, standardized, clinically-labeled yardstick. This is the corner where the capability ledger and the validity ledger come closest to agreeing.
On the protein side that yardstick is ProteinGym: over 250 standardized deep mutational scanning assays comprising more than 2.7 million mutated sequences across more than 200 protein families, plus a clinical benchmark of roughly 65,000 substitution and indel mutations in human genes annotated by domain experts.14 It evaluates over 70 models across zero-shot and supervised settings.14 The motivation is exactly the comparability problem this whole article is about: prior protein predictors were evaluated on distinct, sparse datasets while relative performance fluctuated importantly across assays.14
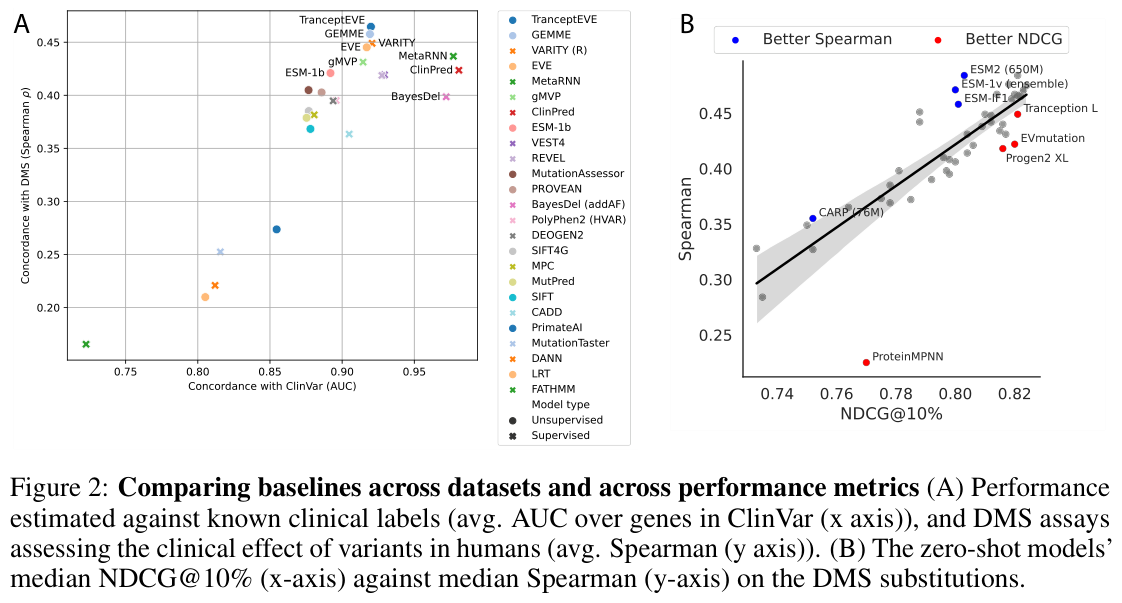 Figure 11: Clinical concordance versus DMS performance for protein-LM variant-effect predictors, the closest the field has to a clinical variant-effect leaderboard. Source: ProteinGym (Notin et al., NeurIPS 2023)
Figure 11: Clinical concordance versus DMS performance for protein-LM variant-effect predictors, the closest the field has to a clinical variant-effect leaderboard. Source: ProteinGym (Notin et al., NeurIPS 2023)
On the DNA side, Evo 2's clinical variant-effect panel is the closest equivalent, spanning ClinVar coding and noncoding variants, splice-altering variants, loss-of-function classification, and the BRCA1 saturation-mutagenesis benchmark.
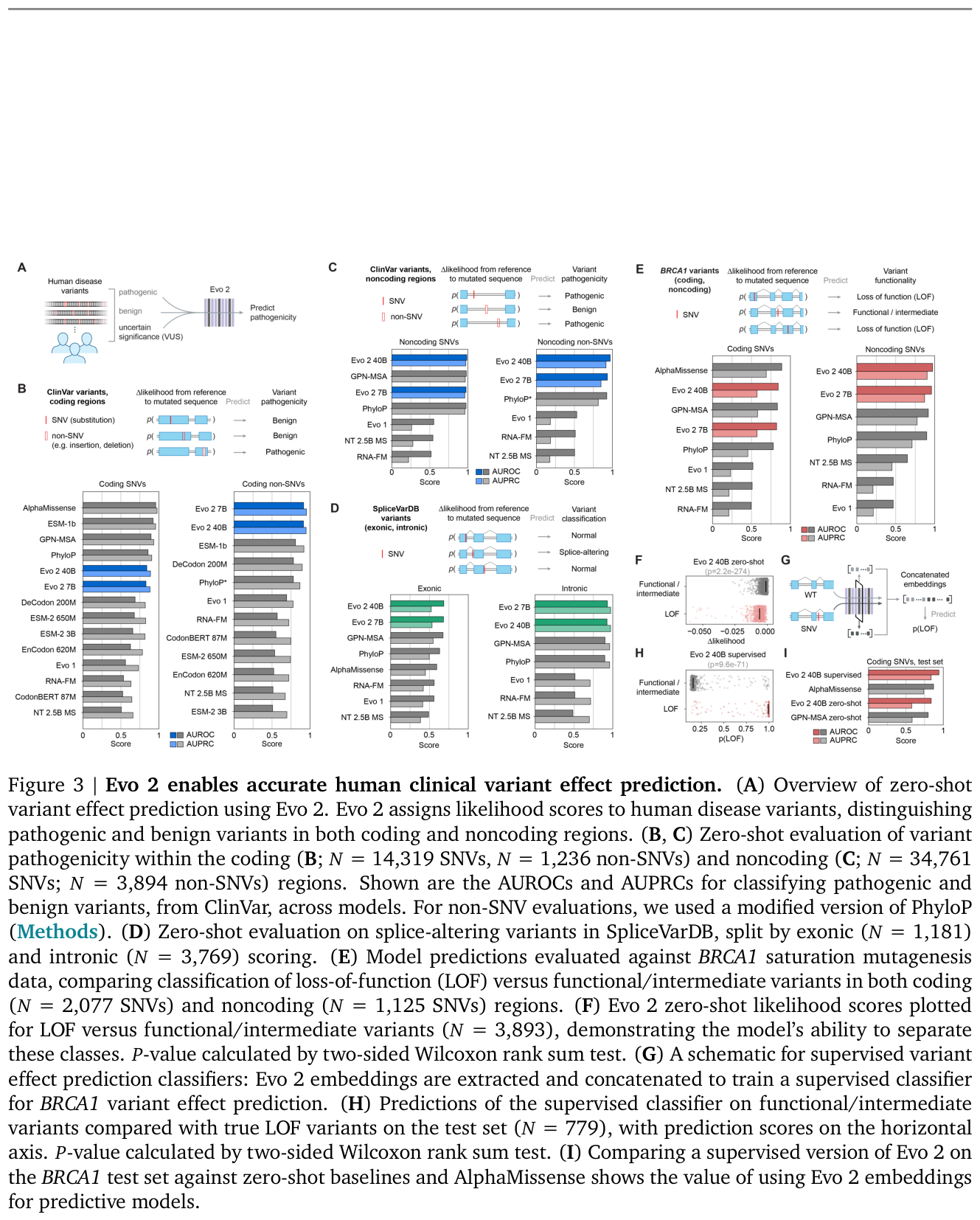 Figure 12: Evo 2 zero-shot clinical variant-effect prediction versus baselines, including the BRCA1 saturation-mutagenesis benchmark. Source: Evo 2 (Brixi et al., Nature 2026)
Figure 12: Evo 2 zero-shot clinical variant-effect prediction versus baselines, including the BRCA1 saturation-mutagenesis benchmark. Source: Evo 2 (Brixi et al., Nature 2026)
Read the two panels together and the calibrated conclusion writes itself. For protein missense interpretation, protein language models are competitive with or ahead of alignment-based methods on standardized clinical labels.14 For DNA, the frontier models are state of the art on noncoding and splicing variants but not on coding SNVs, where specialist tools like AlphaMissense still lead.1 That is the task-dependence GENEB predicts, visible inside a single paper. The right clinical posture is not "use the foundation model" but "use the right model for the variant class, and anchor it to the benchmark that resembles your case." The defensible reading is narrower still: zero-shot variant scores are now good enough to contribute evidence under an ACMG-style framework, especially for noncoding and splice variants where classical tools are weakest, and not good enough to act alone. The maturity here is real, and it is bounded.
Protein and structure models
The protein-structure story is where foundation models first earned the field's trust, and it is also where the speed-versus-accuracy tradeoff is best quantified, because there is independent third-party benchmarking rather than vendor self-report.
ESM-2 and ESMFold made the foundational observation: as protein language models scale from 8 million to 15 billion parameters, an atomic-resolution picture of protein structure emerges in the learned representations, enabling an order-of-magnitude acceleration of high-resolution structure prediction.15 Perplexity falls with scale, with the 8-million-parameter model sitting at 10.45.15 Because ESMFold is MSA-free and works from a single sequence, the speedup over prior pipelines is up to one to two orders of magnitude.15 That speed enabled the ESM Metagenomic Atlas: structures for more than 617 million metagenomic proteins, including over 225 million high-confidence predictions, computed in two weeks on a cluster of 2,000 GPUs.15 Many of those predictions were genuinely novel, with 76.8 percent of high-confidence predictions at least 90 percent distinct from UniRef90 and 12.6 percent lacking any match to experimentally determined structures.15
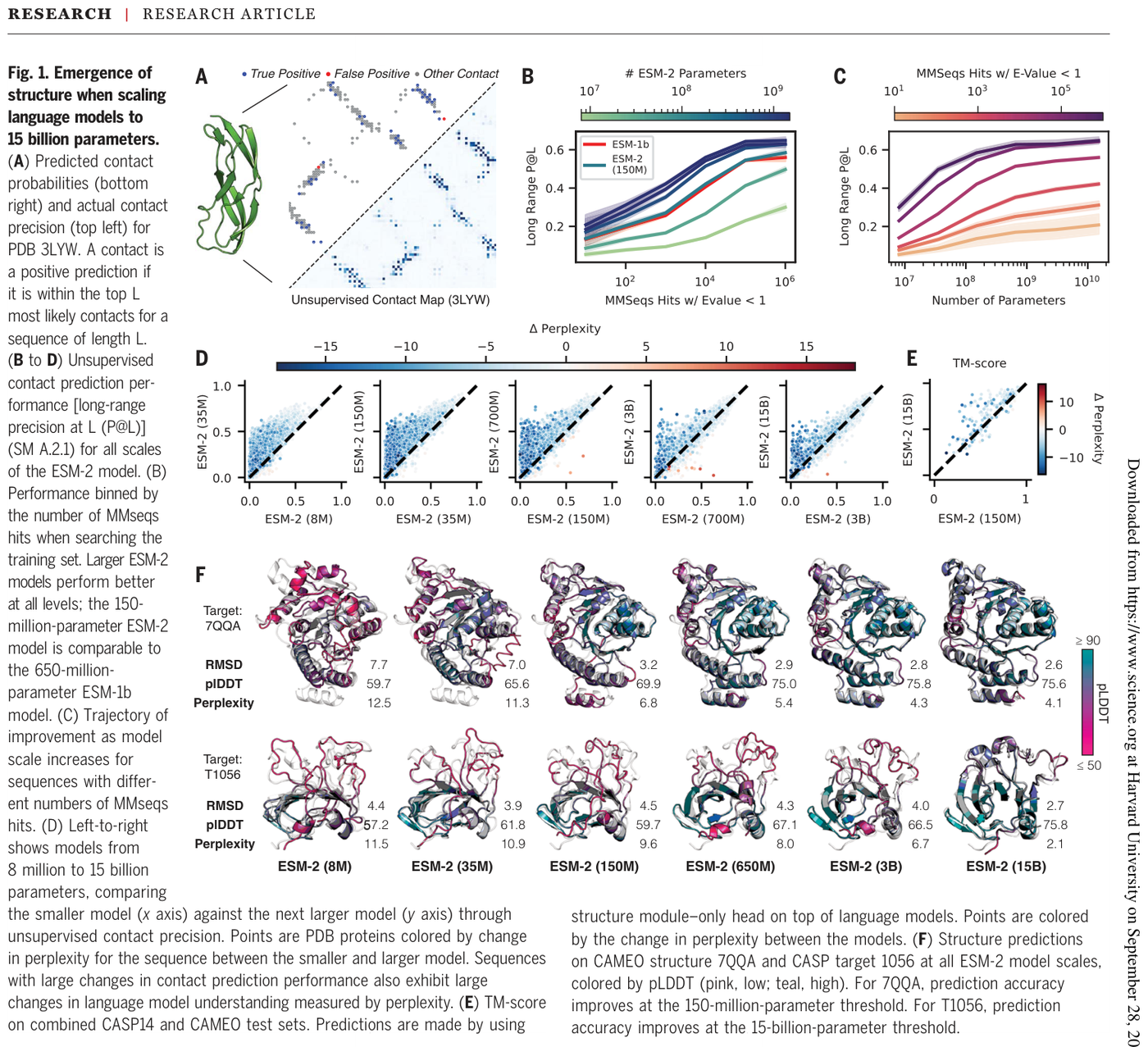 Figure 13: Emergence of protein structure with scale: contact-map precision and example ESMFold predictions improving from 8M to 15B parameters. Source: ESM-2 / ESMFold (Lin et al., Science 2023)
Figure 13: Emergence of protein structure with scale: contact-map precision and example ESMFold predictions improving from 8M to 15B parameters. Source: ESM-2 / ESMFold (Lin et al., Science 2023)
The honest tradeoff comes from an independent 2025-2026 comparison that benchmarked AlphaFold2, AlphaFold3, and ESMFold on deliberately challenging targets: 1,666 monomers and 994 dimers selected at under 40 percent sequence identity and under 70 percent query coverage.16 On those targets, AlphaFold2 and AlphaFold3 correctly predicted 88 percent of monomeric structures and 77 percent of dimeric proteins, while ESMFold predicted 76 percent of monomers and 41 percent of dimers.16 On X-ray and cryo-EM monomers the accuracy was 95 percent for AlphaFold versus 83 percent for ESMFold.16 The summary that survives scrutiny: ESMFold for speed and scale, AlphaFold for accuracy, with the gap widening sharply on complexes.
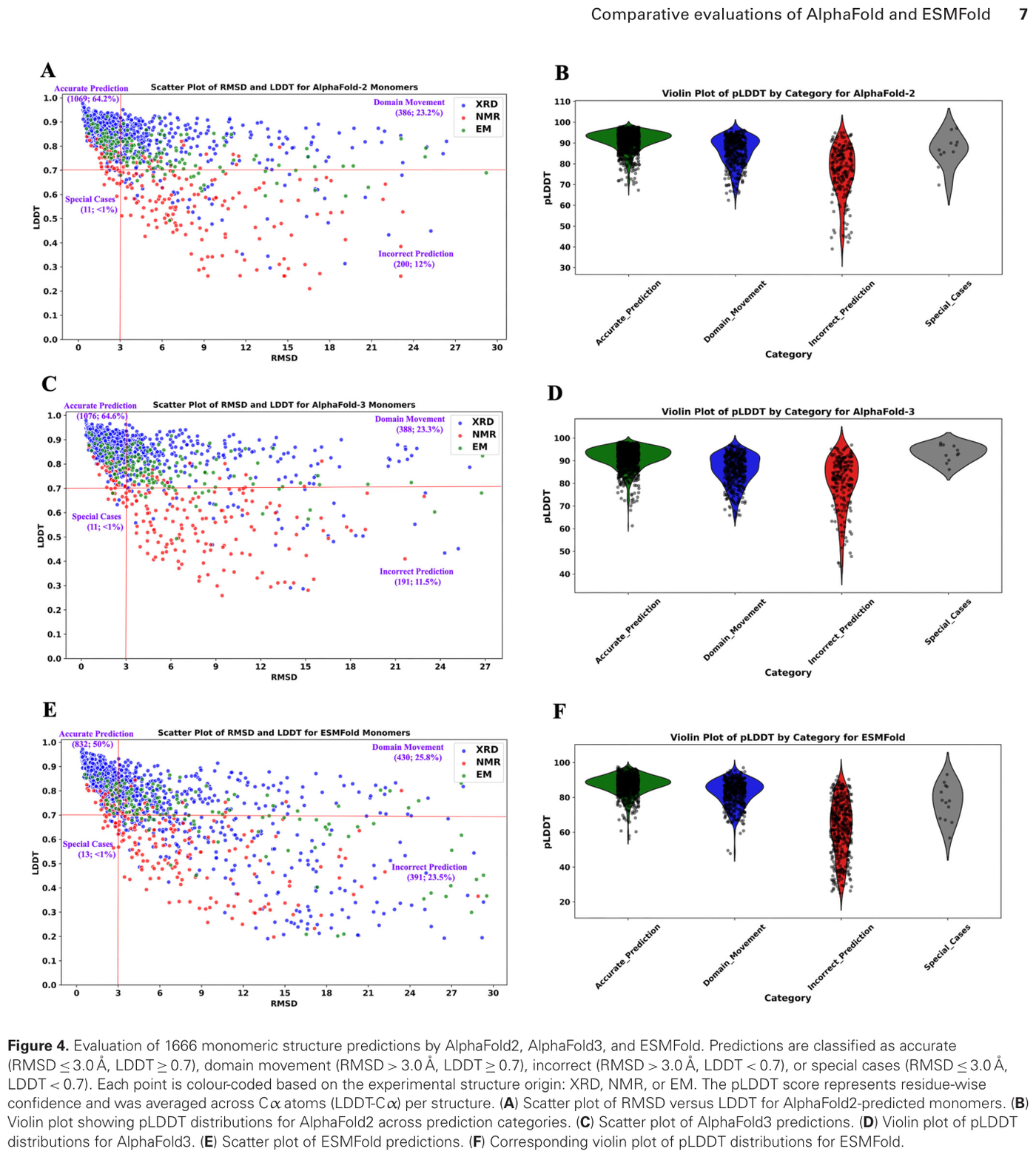 Figure 14: Independent evaluation of monomeric structure predictions by AlphaFold2, AlphaFold3, and ESMFold. Source: ESMFold vs AlphaFold comparison (PMC12809598, 2026)
Figure 14: Independent evaluation of monomeric structure predictions by AlphaFold2, AlphaFold3, and ESMFold. Source: ESMFold vs AlphaFold comparison (PMC12809598, 2026)
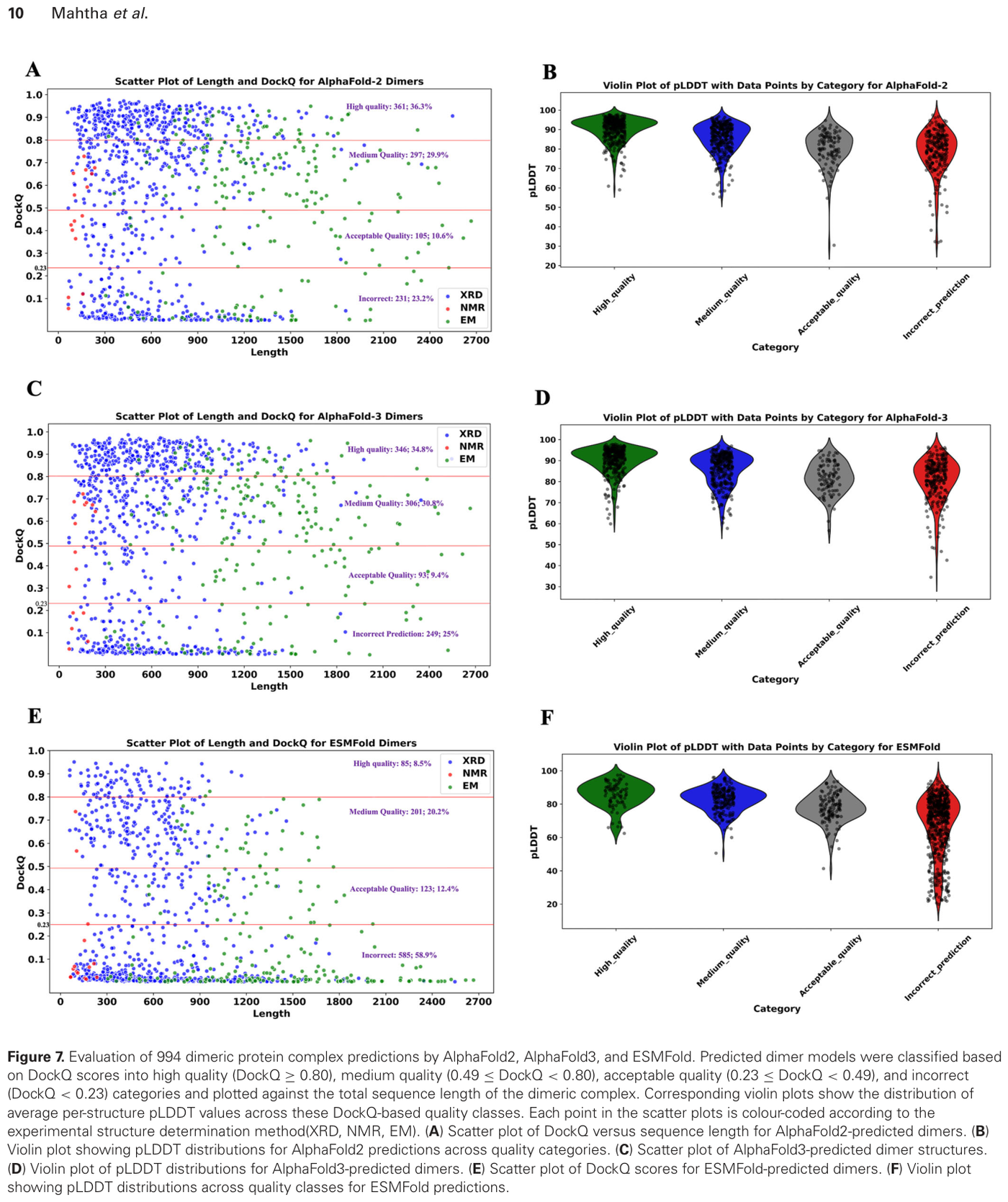 Figure 15: Evaluation of dimeric (protein-protein complex) predictions by AlphaFold2, AlphaFold3, and ESMFold, classified by DockQ accuracy. Source: ESMFold vs AlphaFold comparison (PMC12809598, 2026)
Figure 15: Evaluation of dimeric (protein-protein complex) predictions by AlphaFold2, AlphaFold3, and ESMFold, classified by DockQ accuracy. Source: ESMFold vs AlphaFold comparison (PMC12809598, 2026)
The reason complexes matter is the same reason the field moved to all-atom models. AlphaFold3 extends structure prediction to protein-protein interactions, protein-ligand docking, and protein-nucleic-acid complexes,17 partly by distilling from AF2-generated structures of 41 million MGnify sequences.17 But it carries documented failure modes that a clinical user must know: 4.4 percent of top PoseBusters predictions had a chirality violation, and such violations persisted even when ranking 1,000 predictions with a 100x penalty;17 AF3 may hallucinate structure;17 and antibodies are unreliable because every B cell carries a distinctly shuffled, hypermutated sequence that lacks a usable MSA.17 Because AF3 outputs static structures, disordered regions and multi-state conformations remain hard.17
RoseTTAFold All-Atom is the open, design-capable counterpart. Prior deep-learning structure methods were limited to protein-only systems;18 RFAA combines a residue-based representation of amino acids and DNA bases with an atomic representation of all other groups, modeling assemblies of proteins, nucleic acids, small molecules, metals, and covalent modifications from their sequences and chemical structures.18 Its nucleic-acid predecessor did this by expanding the residue alphabet to 28, covering 20 amino acids, 4 DNA bases, and 4 RNA bases.18 Finetuned for denoising, RFdiffusionAA designs proteins around small molecules, with experimentally validated binders for the cardiac therapeutic digoxigenin, the cofactor heme, and the light-harvesting molecule bilin.18 The honest framing, reported in the AlphaFold 3 work itself, is that an all-atom generalist can model every interaction type but underperforms specialist methods on any single one, while specialist methods simply fail when a complex contains multiple interaction types.17 Generalist versus specialist is the recurring shape of this entire field.
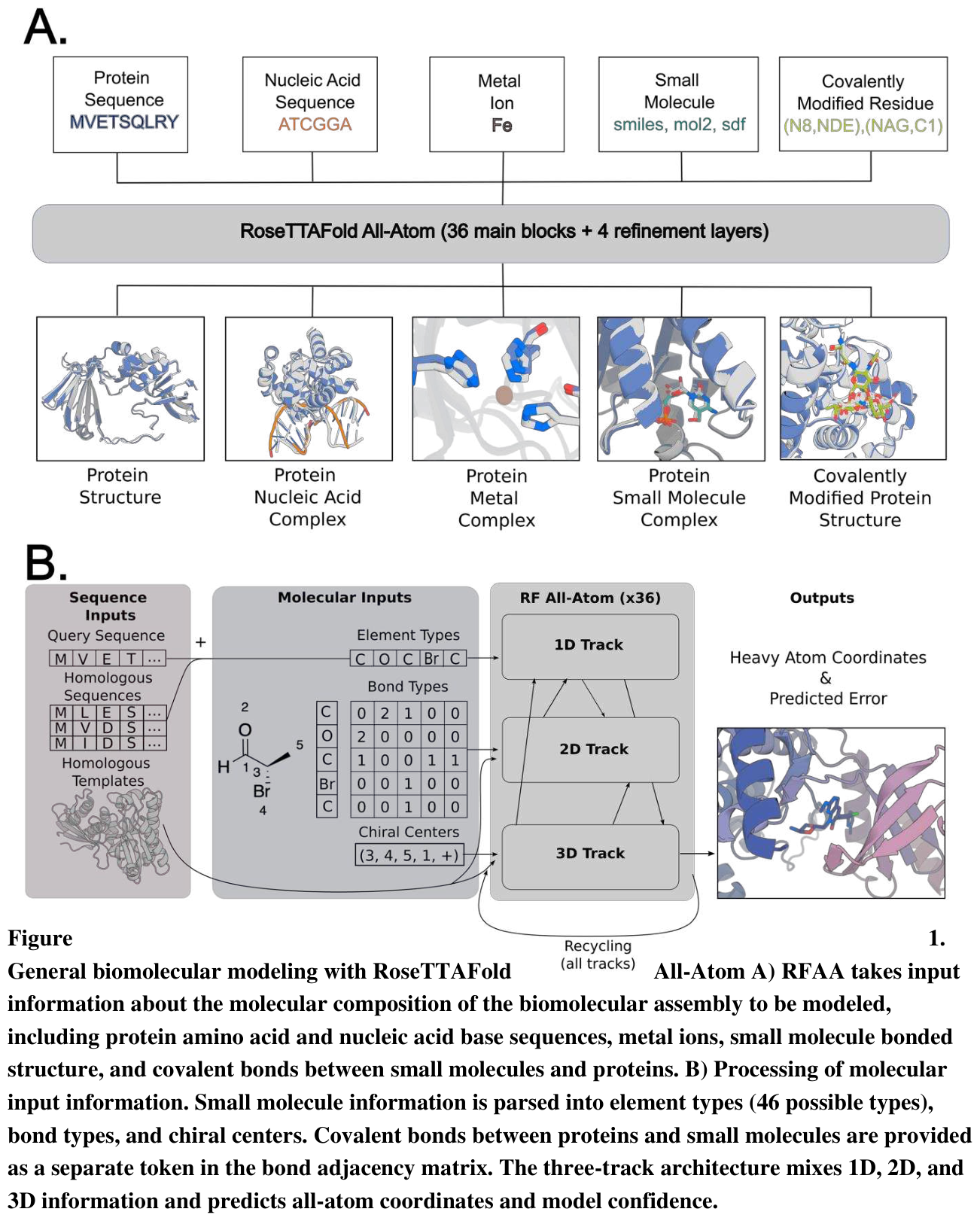 Figure 16: RoseTTAFold All-Atom's general biomolecular representation and three-track architecture. Source: RoseTTAFold All-Atom (Krishna et al., Science 2024)
Figure 16: RoseTTAFold All-Atom's general biomolecular representation and three-track architecture. Source: RoseTTAFold All-Atom (Krishna et al., Science 2024)
ESM3 extends this trajectory toward multimodal generation over protein sequence, structure, and function jointly, which is the direction the protein field is now moving. I am keeping its specific numbers out of this comparison because I could not verify them against a primary archived source, and an unverifiable parameter count has no place in a piece for this audience. Treat it as a qualitative signpost, not a benchmarked entry.
Single-cell and expression models: where the validity ledger is thinnest
Single-cell foundation models are trained at impressive scale. scGPT was pretrained on roughly 33 million non-cancerous human cells;9 Geneformer's current generation is the V2-316M model;19 UCE is a 33-layer model with over 650 million parameters trained on more than 300 datasets totaling over 36 million cells across 8 species, for 40 days on 24 A100 80GB GPUs.20 UCE is interesting architecturally because it represents genes via protein-language-model embeddings to build a universal cross-species latent space and can zero-shot embed cells from species it never saw in training.20 On the capability ledger, these are real and large. The validity ledger is where they thin out, and it is the reason I would not let a single-cell foundation model near a clinical decision today.
The flagship result is from Nature Methods in 2025. Ahlmann-Eltze, Huber, and Anders compared five foundation models and two other deep-learning methods against deliberately simple baselines for predicting transcriptome changes after single or double gene perturbations. None outperformed the baselines.3 For double perturbations, every model had a prediction error substantially higher than a simple additive baseline.3 For single perturbations, none of the deep models consistently beat the mean prediction or the linear model.3 Pretraining on the single-cell atlas provided only a small benefit over random embeddings; only pretraining on perturbation data itself helped.3 The authors' conclusion is the calibrated version of the skeptical case, and it is worth reading aloud: because their deliberately simple baselines cannot represent realistic biological complexity yet were not outperformed, the foundation models' goal of a generalizable representation of cellular states that predicts not-yet-performed experiments is still elusive.3 For context on why this is hard, the same study found only 5,035 genetic interactions out of a potential 124,000 at a 5 percent false discovery rate.3
The zero-shot picture is just as sobering, and zero-shot is the realistic discovery scenario, where you have not finetuned on the answer. Kedzierska and colleagues evaluated Geneformer and scGPT zero-shot and found they may face reliability challenges and can be outperformed by simpler methods.9 Both performed worse than selecting highly variable genes and using established methods like Harmony and scVI for cell-type clustering, as measured by the AvgBio score.9 Highly variable gene selection outperformed Geneformer and scGPT across all metrics.9 They even varied scGPT's pretraining scale across 814,000, 10.3 million, and 33 million cells and still found the only previously-unseen dataset where scGPT beat both baselines was a single PBMC study.9 The structural lesson is the one to keep: strong finetuned benchmark numbers can mask weak general representations, which is why the authors call zero-shot evaluation a critical step before deployment.9
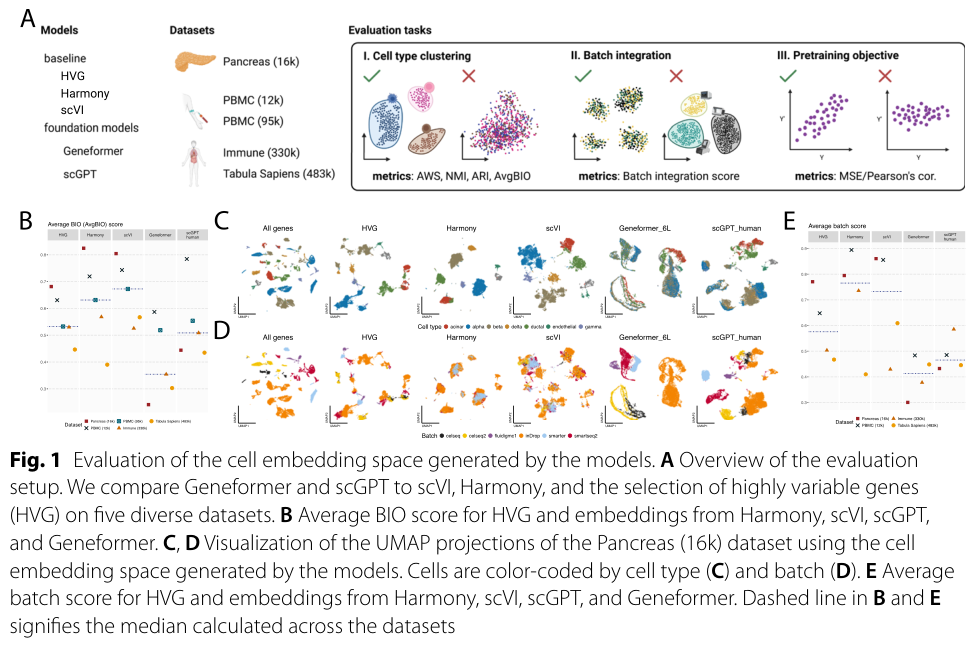 Figure 17: Zero-shot evaluation of the cell-embedding space of Geneformer and scGPT versus simple baselines. Source: Kedzierska et al., Genome Biology 2025
Figure 17: Zero-shot evaluation of the cell-embedding space of Geneformer and scGPT versus simple baselines. Source: Kedzierska et al., Genome Biology 2025
The interpretability claims fare worst of all. A 2026 systematic evaluation built from 37 analyses and 153 statistical tests across four cell types and two perturbation modalities tested whether scGPT and Geneformer attention encodes regulatory biology, as both papers and many downstream studies assume.19 It does not. Attention captures co-expression, not unique regulatory signal: trivial gene-level baselines outperformed both attention and correlation edges (AUROC 0.81 to 0.88 versus 0.70), pairwise edge scores added zero predictive contribution, and causal ablation of the heads claimed to carry regulatory signal produced no degradation at all.19 The specific numbers are stark: trivial gene-level baselines land in the 0.81 to 0.88 AUROC range for predicting CRISPRi targets, with variance alone reaching 0.881, while attention-derived and correlation edges sit near 0.70.19 The attention does encode layer-specific biological structure, but that structure adds no incremental value for the prediction task.19 This matters because both scGPT and Geneformer highlight attention-derived gene network inference as a key application, and downstream studies have adopted attention-derived edge scores as regulatory proxies without rigorous validation.19
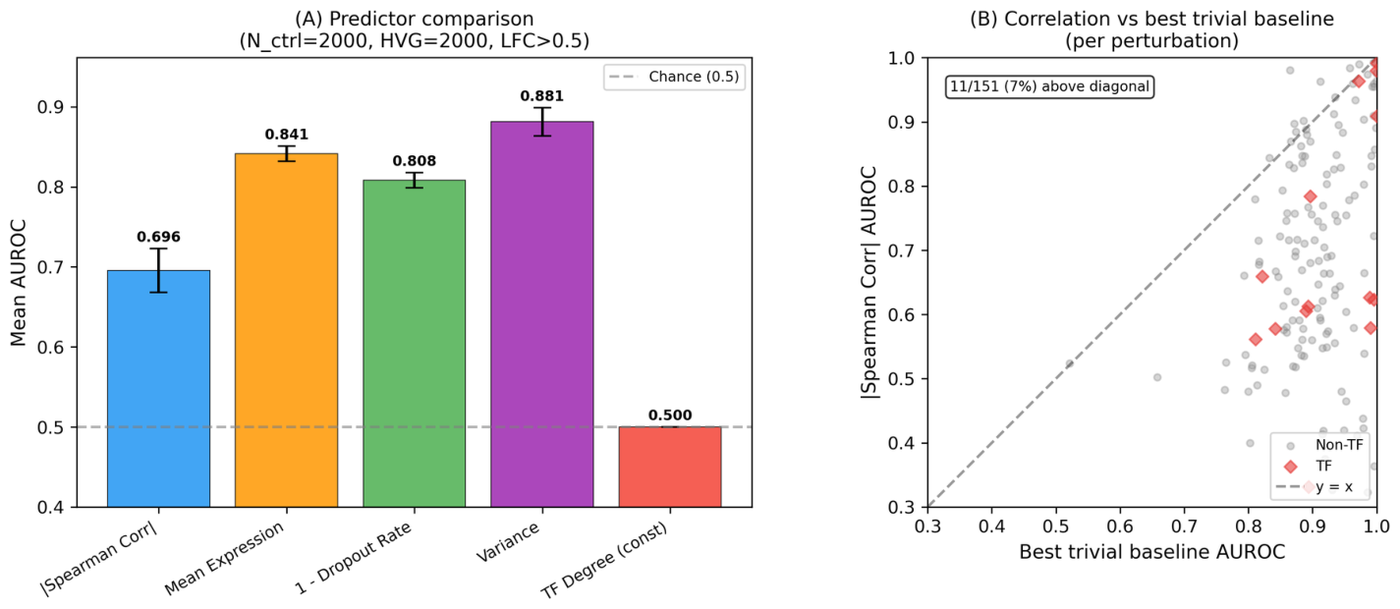 Figure 18: Trivial baselines outperform attention-derived regulatory edges; attention captures co-expression, not regulatory signal. Source: scFM Interpretability Critique (arXiv:2602.17532, 2026)
Figure 18: Trivial baselines outperform attention-derived regulatory edges; attention captures co-expression, not regulatory signal. Source: scFM Interpretability Critique (arXiv:2602.17532, 2026)
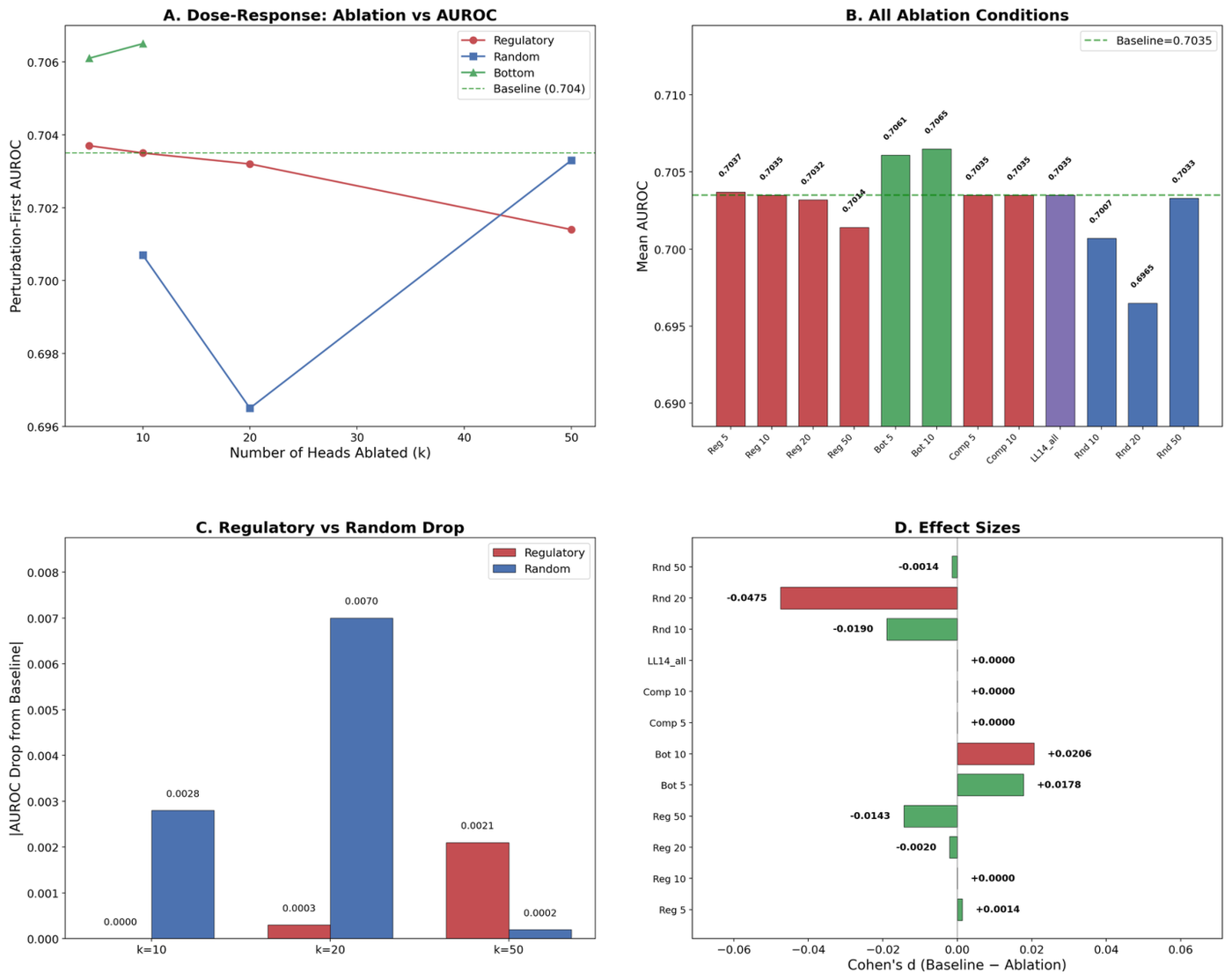 Figure 19: Causal ablation of attention heads claimed to carry regulatory signal produces no degradation. Source: scFM Interpretability Critique (arXiv:2602.17532, 2026)
Figure 19: Causal ablation of attention heads claimed to carry regulatory signal produces no degradation. Source: scFM Interpretability Critique (arXiv:2602.17532, 2026)
To be fair to the field, the same critique offers a constructive fix, Cell-State Stratified Interpretability, which improves gene-regulatory-network recovery up to 1.85x by addressing an attention-specific scaling failure.19 And the peer-reviewed reviews are candid about the open problems: single-cell foundation models face the nonsequential nature of omics data, inconsistent data quality, heavy compute demands, and the difficulty of interpreting latent embeddings.21 The point is not that these models are useless. The point is that, for a clinical audience in 2026, the published evidence does not support using single-cell foundation model attention as a regulatory-network oracle, nor using these models for perturbation prediction where a linear baseline is cheaper and at least as good.
Multimodal directions
The genuinely new direction since the January guide is multimodal biomolecule modeling, and it takes three distinct forms worth separating. All-atom complex models (AlphaFold3, RoseTTAFold All-Atom) unify molecule types in a single structural prediction.1817 Sequence-structure-function generative models (ESM3) unify representation and generation within the protein modality. Cross-modal embedding (UCE representing genes through protein-LM embeddings) borrows one modality's learned space to ground another.20 A truly unified DNA-plus-protein-plus-expression clinical model does not exist yet; what exists are bridges between modalities, each useful in its niche and none yet validated as a diagnostic. The 2025 systematic review of generative AI in medical genetics (195 studies under PRISMA 2020) makes the same point from the clinical side: transformer-based models perform well on tasks like variant interpretation and tentative molecular diagnosis, but major challenges persist in integrating genomic sequence, imaging, and clinical records into unified clinical pipelines that hold up in practice, with real limits on generalizability.22
Where this comparison is unfair
I have argued for a divided verdict: mature for variant effect prediction, not for perturbation or interpretability. A skeptical reader should attack the framing I have built, so let me do it first, on five fronts.
The variant-effect maturity claim leans on retrospective benchmarks that may be contaminated. Evo 2's BRCA1 result and AlphaGenome's 24-of-26 are real, but they are discrimination on labeled sets that overlap the training distribution.12 I have no prospective, ancestry-stratified, contamination-controlled clinical trial to point to, and neither does anyone else. The maturity I am claiming is benchmark maturity, not demonstrated prospective clinical utility, and those are not the same thing.
Contamination cuts both ways, though. I flagged train and test leakage as a reason to distrust high benchmark numbers, but the strongest clinical signals are partly insulated from that critique because they are scored against experimental or expert-curated labels rather than purely against held-out reference sequence. Evo 2 on BRCA1 noncoding is evaluated against saturation-mutagenesis data,1 AlphaGenome on splicing against measured splicing outcomes,2 and protein language models against ProteinGym's expert-annotated clinical labels.14 The skepticism should be calibrated to what each benchmark actually measures, not applied as a blanket discount.
The skeptical results are themselves task-specific and could be over-generalized. The Nature Methods negative result is about perturbation prediction; the interpretability critique is about attention-as-regulatory-network; the zero-shot critique is about clustering and integration.3199 None of them shows that single-cell foundation models are worthless for, say, supervised cell-type annotation after finetuning. Using them as a blanket dismissal would be exactly the inverted-hype failure mode this audience distrusts. A reader should weight the single-cell critiques most heavily against single-cell claims.
GENEB's probing protocol understates fine-tuned models, and its instability cuts both ways. GENEB evaluates frozen representations,4 which can make a model built to be fine-tuned look mediocre as a feature extractor. And if leaderboards are unstable and rankings flip by task category, then any single comparison, including the favorable variant-effect ones, is contingent on the task set chosen. The same evidence that lets me discount inflated leaderboard wins also forbids me from over-trusting the wins I happen to like. The category-level view, not the aggregate, is the only defensible one.
Open versus API-only is a values judgment, not a benchmark. AlphaGenome's 24-of-26 result stands on its merits;2 my discomfort with API-only access is a deployment and reproducibility concern for regulated diagnostics, not a claim that the model is less accurate. A research group with no validation-freezing requirement should weigh it differently than a clinical lab. And one more honest caveat: the newest models, Evo 2, AlphaGenome, and GENEB itself, are 2026 work that has not yet faced the years of adversarial replication the structure-prediction comparisons have. Some of my confidence ordering reflects how much scrutiny each result has survived, which is itself a moving target.
A decision framework for the clinic
The practical consequence of leaderboard instability is that model selection has to be driven by task category, not by a single winner. Here is the decision logic I would apply, expressed as a tree, followed by a matrix. Read both as scope-limited recommendations for evaluation and research, not as endorsements for unsupervised clinical use.
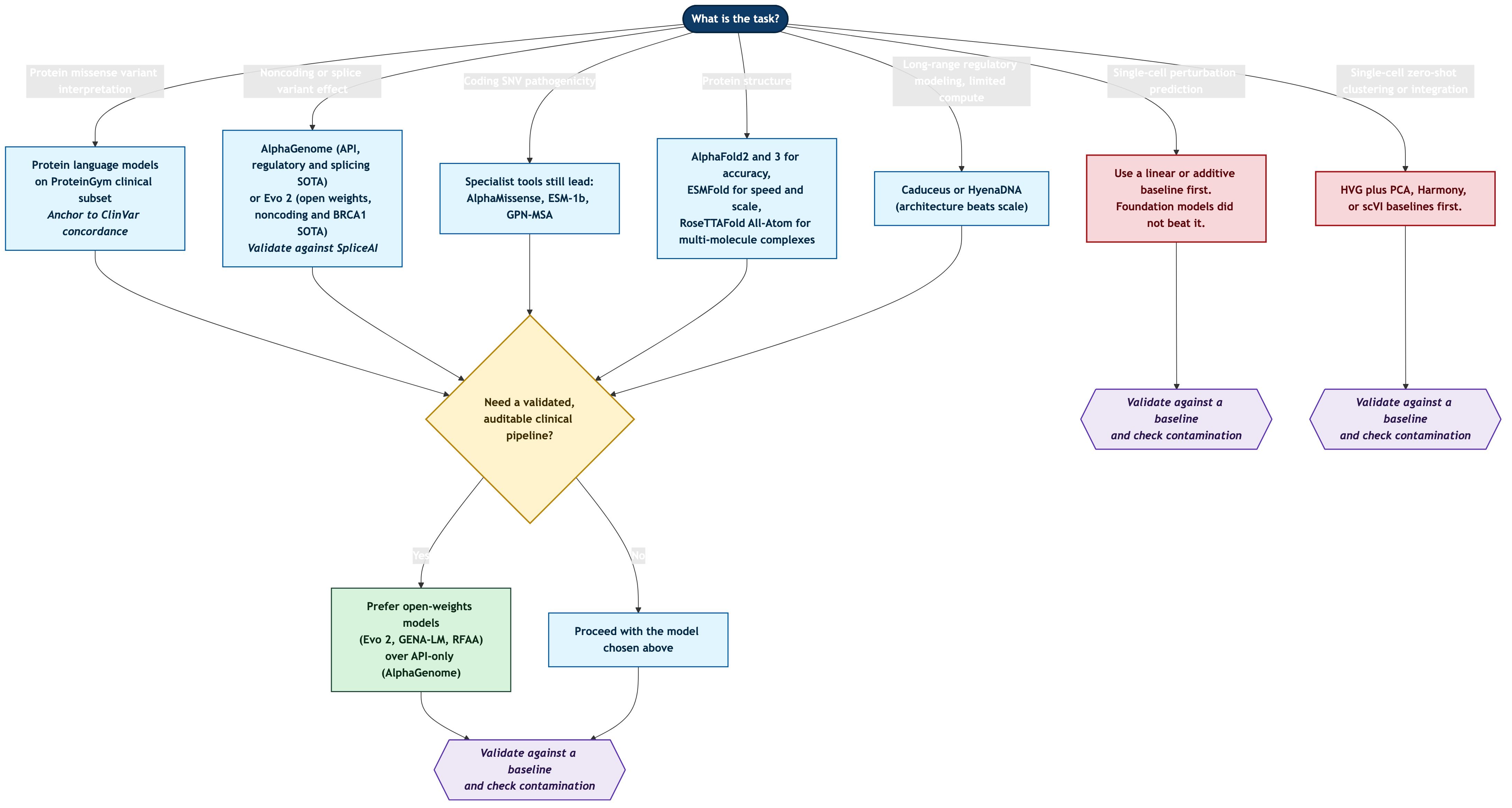 Figure 20: Decision logic for model selection by task category. Recommendations are starting points for evaluation against a baseline, not endorsements for unsupervised clinical use.
Figure 20: Decision logic for model selection by task category. Recommendations are starting points for evaluation against a baseline, not endorsements for unsupervised clinical use.
The matrix below condenses the same logic. Each row names the honest baseline a model has to beat before you trust it, and the caveat that travels with the recommendation.
| Task | Reasonable first choice in 2026 | Honest baseline to beat first | The caveat that travels with it |
|---|---|---|---|
| Noncoding / splice variant effect | AlphaGenome; Evo 2 | SpliceAI and prior regulatory predictors | Retrospective benchmarks; AlphaGenome is API-only and non-commercial2 |
| Coding SNV pathogenicity | Specialist tools (AlphaMissense, ESM-1b, GPN-MSA) | A calibrated specialist missense tool | Evo 2 ranked 4th/5th on coding SNVs behind these1 |
| Protein missense interpretation | Protein language models on ProteinGym tracks | EVmutation / alignment-based predictors | Relative ranking fluctuates across assays14 |
| Protein structure (accuracy) | AlphaFold2/3 | A specialist where a single interaction dominates | AF3 chirality violations (4.4%), hallucination, weak antibodies17 |
| Protein structure (speed/scale) | ESMFold | AlphaFold2 where accuracy is paramount | 76% monomer / 41% dimer vs 88% / 77% on hard targets16 |
| Multi-molecule complexes / design | RoseTTAFold All-Atom; AlphaFold3 | Specialist docking for single-interaction cases | Generalists underperform specialists per interaction type17 |
| Long-range regulatory, limited compute | Caduceus; HyenaDNA | A well-tuned CNN on Genomic Benchmarks | Caduceus beats 10x larger models8; still benchmark-stage for clinical use |
| Single-cell perturbation prediction | Linear / additive baseline first | The additive / linear baseline (likely wins) | No FM beat simple baselines in any setting tested3 |
| Single-cell zero-shot clustering | HVG + PCA / Harmony / scVI | HVG + PCA, Harmony, scVI | HVG outperformed Geneformer and scGPT across all metrics9 |
| Regulatory network inference from attention | Do not rely on attention edges | Co-expression / mean-expression baseline | Trivial baselines 0.81-0.88 vs attention 0.70; ablation no degradation19 |
Table 1: A task-driven selection matrix. Every recommendation is a starting point for evaluation against a baseline on your own data, not an endorsement for unsupervised clinical use.
Two operating principles run through the whole table. Anchor every model to a baseline on your own data before you trust it, because the field's clearest lesson is that baselines win more often than the marketing admits. And prefer open, version-lockable models for anything approaching a clinical pipeline, because reproducibility under a quality system is not optional. The right first experiment makes both habits concrete: run a frozen open model and a trivial baseline on the same split, and report them side by side so the model only counts as useful if it actually beats the baseline.
# variant_effect_quickstart.py (condensed; full tested version in repo)
# The habit that matters: a foundation model only counts if it beats a trivial baseline.
# Pinned: torch==2.5.1 transformers==4.46.3 scikit-learn==1.9.0 biopython==1.87
import numpy as np
from Bio.Align import substitution_matrices
from sklearn.metrics import roc_auc_score
ESM2 = "facebook/esm2_t12_35M_UR50D"
ESM2_REV = "6fbf070e65b0b7291e7bbcd451118c216cff79d8" # pin the commit, not just the tag
def score_esm2(variants):
"""Zero-shot masked-marginal LLR: log P(wt) - log P(mut). Higher = more damaging."""
import torch
from transformers import AutoModelForMaskedLM, AutoTokenizer
tok = AutoTokenizer.from_pretrained(ESM2, revision=ESM2_REV)
model = AutoModelForMaskedLM.from_pretrained(ESM2, revision=ESM2_REV).eval()
out = []
with torch.no_grad():
for v in variants:
ids = tok(v.sequence, return_tensors="pt")["input_ids"].clone()
i = v.position # leading <cls> shifts 1-based position to this index
ids[0, i] = tok.mask_token_id
lp = torch.log_softmax(model(input_ids=ids).logits[0, i], dim=-1)
wt, mut = tok.convert_tokens_to_ids(v.wt_aa), tok.convert_tokens_to_ids(v.mut_aa)
out.append(float(lp[wt] - lp[mut]))
return np.asarray(out)
def score_blosum62(variants):
"""Trivial baseline: negated BLOSUM62 substitution score. No model, no GPU."""
b = substitution_matrices.load("BLOSUM62")
return np.asarray([-float(b[v.wt_aa, v.mut_aa]) for v in variants])
def auroc_ci(labels, scores, n_boot=2000, seed=0):
"""AUROC with a percentile bootstrap 95% CI."""
rng = np.random.default_rng(seed)
point = roc_auc_score(labels, scores)
boots = []
for _ in range(n_boot):
idx = rng.integers(0, len(labels), len(labels))
if len(np.unique(labels[idx])) == 2:
boots.append(roc_auc_score(labels[idx], scores[idx]))
lo, hi = np.percentile(boots, [2.5, 97.5])
return point, lo, hi
# variants: a HELD-OUT, ancestry-stratified ClinVar pathogenic-vs-benign split.
# CONTAMINATION CAVEAT: ESM-2 trained on UniRef50; ClinVar overlaps heavily, so a
# high AUROC is partly memorization. Treat it as an upper bound, not a deployment number.
# m = auroc_ci(labels, score_esm2(variants)); b = auroc_ci(labels, score_blosum62(variants))
# The model "passes" only if it clears the baseline AUROC and the bootstrap CIs separate.
# The same harness applies to DNA models
# (Caduceus, Evo 2) by swapping score_esm2 for their per-token log-likelihood API.
# Representative output (illustrative; always judge on your own held-out split).
# Case 1, the model earns its place:
ESM-2 (esm2_t12_35M_UR50D) AUROC = 0.93 95% CI [0.90, 0.95]
BLOSUM62 baseline (trivial) AUROC = 0.82 95% CI [0.77, 0.86] -> PASS (clears baseline, intervals separate)
# Case 2, the model adds nothing the baseline did not:
ESM-2 (esm2_t12_35M_UR50D) AUROC = 0.84 95% CI [0.79, 0.89]
BLOSUM62 baseline (trivial) AUROC = 0.83 95% CI [0.78, 0.88] -> FAIL (intervals overlap)
What is settled, what is not, and what would change my mind
What the evidence supports today: protein and DNA foundation models are the right tool for variant effect prediction at the benchmark level, with protein-LM missense interpretation anchored to ProteinGym's clinical subset14 and DNA models leading on noncoding and splicing variants12 while ceding coding SNVs to specialists.1 Structure prediction is mature, with a well-quantified accuracy-versus-speed frontier between AlphaFold and ESMFold16 and an all-atom generalization frontier between AlphaFold3 and RoseTTAFold All-Atom.1817 Architecture and pretraining alignment matter more than parameter count, which is why a reverse-complement-equivariant Caduceus can beat models ten times its size.84
What the evidence does not yet support: single-cell foundation models for perturbation prediction, where they lose to linear baselines;3 their attention as a regulatory-network oracle, where trivial baselines win and causal ablation finds nothing;19 their zero-shot embeddings over simple HVG pipelines;9 and, critically, any frontier model as a validated, prospective diagnostic. Retrospective AUROC is not prospective clinical utility, API-only access is a reproducibility liability for a regulated lab, and the standardized benchmarks themselves are unstable enough that the same model can be a breakthrough in one paper and an underperformer in another.4
What would change my mind, and what I would ask the Nature-published colleagues I am sharing a stage with to produce: a prospective, ancestry-stratified, contamination-controlled evaluation of a frozen, openly available variant-effect model against the specialist tool it claims to beat and against a deliberately simple baseline, reporting concordance with eventual clinical classification rather than retrospective ClinVar discrimination, with confidence intervals and a preregistered protocol. Genomics has spent a decade arguing about the ruler.12 The day we agree on one, and a foundation model wins on it fairly, is the day the variant-effect corner of the capability ledger finally earns its place on the validity ledger too. Until then, the correct stance for molecular pathology is to use these models where the benchmark evidence is strong and the access is open, to baseline everything, and to treat every leaderboard claim as a hypothesis about your data rather than a result on it. The capability curve is steep and real. The validity curve is flatter than the press releases, and in 2026 the gap between them is exactly where clinical judgment has to live.
References
Note on scGPT: the scGPT pretraining scale of roughly 33 million non-cancerous human cells is cited here to the zero-shot evaluation source9, where it is reported as part of that study's scale ablation. The primary reference is Cui H, Wang C, Maan H, et al. scGPT: toward building a foundation model for single-cell multi-omics using generative AI. Nature Methods 2024;21:1470-1480. https://www.nature.com/articles/s41592-024-02201-0
Reproducibility note
All quantitative claims in this article are direct, verified quotes from the primary sources cited inline, drawn from a verified-fact set in which each statistic was checked against the original paper at the quoted location. Figures are reproduced from the cited papers and retain their source attribution; every figure carrying a quantitative claim keeps a citation in its caption. Where a model is API-only (AlphaGenome) or server-gated (AlphaFold3), the access constraint is stated in text because it bears directly on clinical reproducibility. The quickstart code placeholder is designed to be run against an in-house ClinVar split with a trivial BLOSUM62 baseline included and an exact model revision pinned, so that any reported model advantage is reproducible and baseline-anchored on the reader's own data.
This article deliberately does not make three claims that the underlying sources do not support, and a skeptical reader should hold me to their absence:
- No specific numeric BRCA1 accuracy. The verified result is that Evo 2 "set a new state-of-the-art for BRCA1 noncoding SNVs"1 without task-specific finetuning. I do not report any percentage accuracy for that benchmark, because none appears in the verified source text; a figure of that kind circulating in summaries is not traceable to the primary result.
- No ancestry-stratified performance figure. None of the cited papers reports variant-effect performance broken down by genetic ancestry, so I make no claim about how these models perform across non-European populations. That gap is itself a reason for clinical caution.
- No prospective decision-impact metric. Every clinical-sounding number here is retrospective discrimination on curated labels. I make no claim that any model has been shown to change a clinical decision prospectively, because no such study exists in this evidence set.
Footnotes
-
Brixi G, Durrant MG, Ku J, et al. Genome modelling and design across all domains of life with Evo 2. bioRxiv 2025.02.18.638918; Nature 2026. https://www.biorxiv.org/content/10.1101/2025.02.18.638918v1 ; https://www.nature.com/articles/s41586-026-10176-5 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20 ↩21 ↩22 ↩23
-
Avsec Z, et al. Advancing regulatory variant effect prediction with AlphaGenome. bioRxiv 2025.06.25.661532; Nature 2026. https://www.biorxiv.org/content/10.1101/2025.06.25.661532v1 ; https://www.nature.com/articles/s41586-025-10014-0 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15
-
Ahlmann-Eltze C, Huber W, Anders S. Deep-learning-based gene perturbation effect prediction does not yet outperform simple linear baselines. Nature Methods 2025. https://www.nature.com/articles/s41592-025-02772-6 (bioRxiv 2024.09.16.613342; PMC12328236) ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10
-
Ledneva D, Nuridinov M, Kuznetsov D. GENEB: Why Genomic Models Are Hard to Compare. arXiv 2026. arXiv:2606.04525. https://arxiv.org/abs/2606.04525 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12
-
Nguyen E, Poli M, Faizi M, et al. HyenaDNA: Long-Range Genomic Sequence Modeling at Single Nucleotide Resolution. NeurIPS 2023. arXiv:2306.15794. https://arxiv.org/abs/2306.15794 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8
-
Nguyen E, Poli M, Durrant MG, et al. Sequence modeling and design from molecular to genome scale with Evo. Science 2024. https://arcinstitute.org/manuscripts/Evo.pdf ↩ ↩2
-
Zhou Z, Ji Y, Li W, Dutta P, Davuluri RV, Liu H. DNABERT-2: Efficient Foundation Model and Benchmark for Multi-Species Genomes. ICLR 2024. arXiv:2306.15006. https://arxiv.org/abs/2306.15006 ↩ ↩2 ↩3 ↩4 ↩5 ↩6
-
Schiff Y, Kao C-H, Gokaslan A, Dao T, Gu A, Kuleshov V. Caduceus: Bi-Directional Equivariant Long-Range DNA Sequence Modeling. ICML 2024. arXiv:2403.03234. https://arxiv.org/abs/2403.03234 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7
-
Kedzierska KZ, et al. Zero-shot evaluation reveals limitations of single-cell foundation models. Genome Biology 2025. PMC12007350. https://pmc.ncbi.nlm.nih.gov/articles/PMC12007350/ ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11
-
Marin FI, Teufel F, Horlacher M, et al. BEND: Benchmarking DNA Language Models on Biologically Meaningful Tasks. ICLR 2024. arXiv:2311.12570. https://arxiv.org/abs/2311.12570 ↩ ↩2 ↩3
-
OmniGenBench: Automating Large-scale in-silico Benchmarking for Genomic Foundation Models. arXiv 2024. arXiv:2410.01784. https://arxiv.org/abs/2410.01784 ↩
-
Gresova K, Martinek V, Cechak D, Simecek P, Alexiou P. Genomic benchmarks: a collection of datasets for genomic sequence classification. BMC Genomic Data 2023;24:25. PMC10150520. https://pmc.ncbi.nlm.nih.gov/articles/PMC10150520/ ↩ ↩2 ↩3
-
Fishman V, Kuratov Y, et al. GENA-LM: a family of open-source foundational DNA language models for long sequences. Nucleic Acids Research 2025;53(2):gkae1310. https://academic.oup.com/nar/article/53/2/gkae1310/7954523 ↩ ↩2
-
Notin P, Kollasch A, Ritter D, et al. ProteinGym: Large-Scale Benchmarks for Protein Fitness Prediction and Design. NeurIPS 2023. PMC10723403. https://papers.nips.cc/paper_files/paper/2023/file/cac723e5ff29f65e3fcbb0739ae91bee-Paper-Datasets_and_Benchmarks.pdf ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7
-
Lin Z, Akin H, Rao R, Hie B, et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023;379:1123-1130. https://www.science.org/doi/10.1126/science.ade2574 ↩ ↩2 ↩3 ↩4 ↩5
-
Comparative evaluation of the prediction accuracy of AlphaFold and ESMFold for monomeric and dimeric proteins. NAR Genomics and Bioinformatics 2026;8:lqag002. PMC12809598. https://pmc.ncbi.nlm.nih.gov/articles/PMC12809598/ ↩ ↩2 ↩3 ↩4 ↩5
-
Abramson J, Adler J, Dunger J, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024;630:493-500. https://www.nature.com/articles/s41586-024-07487-w (PMC11168924) ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11
-
Krishna R, Wang J, Ahern W, et al. Generalized biomolecular modeling and design with RoseTTAFold All-Atom. Science 2024;384:eadl2528. https://www.science.org/doi/10.1126/science.adl2528 ↩ ↩2 ↩3 ↩4 ↩5 ↩6
-
Systematic Evaluation of Single-Cell Foundation Model Interpretability. arXiv 2026. arXiv:2602.17532. https://arxiv.org/pdf/2602.17532 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10
-
Rosen Y, Roohani Y, Agarwal A, et al. Universal Cell Embeddings: A Foundation Model for Cell Biology. bioRxiv 2023. https://www.biorxiv.org/content/10.1101/2023.11.28.568918 ↩ ↩2 ↩3
-
Single-cell foundation models: bringing artificial intelligence into cell biology. 2025. PMC12586647. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12586647/ ↩
-
A systematic review on the generative AI applications in human medical genetics. Frontiers in Genetics 2025. PMC12863965. https://pmc.ncbi.nlm.nih.gov/articles/PMC12863965/ ↩