Carbon-3B matches Evo2-7B on sequence recovery, variant-effect prediction, and motif-perturbation discrimination while generating DNA over 150 times faster.12 It does this with less than half the parameters, a stock Llama-style decoder, and a tokenizer that throws away single-nucleotide resolution at the input layer, then claws it back at the loss.2
That last clause is the whole story. The Carbon team did not invent a new attention operator or a bidirectional state-space block. They took the boring modern LLM stack and asked which parts of the recipe assume natural language, and what breaks when you feed it eukaryotic DNA instead. The answer was tokenization and the loss function. Fix those two, run the rest unchanged, and a relatively standard Transformer holds its own against models with longer training contexts and more parameters.2
The speed gap is not a footnote. It is the point.
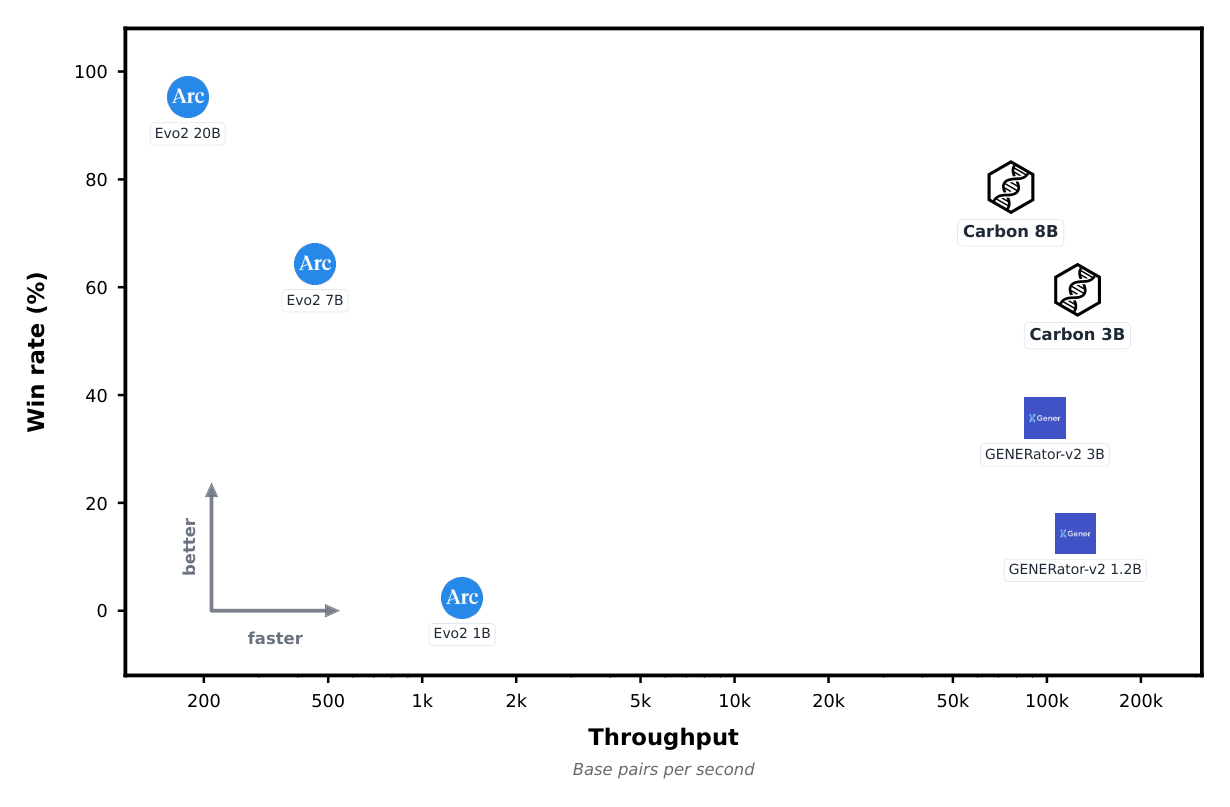
Figure 1: Win rate across seven zero-shot DNA benchmarks versus throughput (base pairs per second, log scale). Carbon models match or beat Evo2-7B at over 150x the speed. Win rates are computed across seven zero-shot DNA benchmarks; throughput is measured over 256 DNA sequences with 1080 bp for prefill and decode. Source: Carbon Technical Report (Hugging Face Biology, 2025).2
For a computational biologist, the practical reading of Figure 1 is simple. If you want to score every variant in a panel, generate candidate regulatory sequences, or run retrieval over genome-scale windows, throughput decides whether the experiment is feasible this week or this quarter. Carbon-3B was built to move that constraint. The models, data, training code, and evaluation suite are all open, including new training-free probes for sequence-level perturbation and long-context retrieval.2 Carbon is a joint project of Hugging Face, Zhongguancun Academy, and TIGEM / University of Naples Federico II.1
The problem: resolution fights context
A genomic foundation model has to satisfy two requirements that pull in opposite directions. The Carbon authors state it plainly: a central challenge in DNA modeling is reconciling single-nucleotide resolution with long-context reasoning.2 Variant-effect prediction has to resolve a single base. A pathogenic ClinVar variant and its benign neighbor differ by one nucleotide, so the model has to score at that resolution. Regulatory reasoning needs the opposite. Enhancers, insulators, and distal regulatory elements sit tens or hundreds of kilobases from the genes they control, so the model has to carry context across that span.
DNA also resists the assumptions baked into language-model recipes. Genomic sequences are "noisy, redundant, sparsely constrained, unevenly annotated, and shaped by evolutionary rather than communicative pressures."2 In humans the genome spans roughly 3 billion base pairs, yet only about 1 to 2 percent directly codes for protein, and roughly 50 percent consists of repetitive sequence derived from ancient transposable elements.2 The authors call this a low signal-to-noise ratio of raw DNA.2 Train on raw genome and most of the gradient goes into modeling repeats and intergenic filler.
The most direct way to preserve single-nucleotide resolution is single-nucleotide tokenization, where each base is one token.2 Evo2 takes this route, and it is a strong reference point for single-nucleotide modeling.23 The cost is sequence length. For a Transformer, attention scales quadratically with token length, so increasing context under single-nucleotide tokenization quickly becomes expensive.2 A 393 kbp window at one base per token is 393,000 positions of quadratic attention, which is the root of Evo2's throughput problem in Figure 1. You can buy long context by abandoning attention entirely, the path HyenaDNA and Caduceus take with long-convolution and state-space operators, but those models give up the mature optimization and serving ecosystem built around attention.245
Carbon goes the other way. It keeps a standard decoder-only Transformer and reconciles the tension by separating where resolution lives. Resolution does not have to live in the tokenizer. It can live in the loss. That reframing runs through every design choice that follows: the tokenizer compresses sequence length, the data pipeline raises signal density, and the loss supervises at the base level even when the output vocabulary is coarser.2
The architecture: Llama 3, deliberately boring
Carbon-3B is a 3B-parameter decoder-only autoregressive genomic foundation model trained on DNA and RNA, with a primary focus on eukaryotes.1 The backbone is a standard decoder-only Transformer based on Llama 3: a pre-norm causal Transformer with RMSNorm, SwiGLU feed-forward blocks, rotary position embeddings, and grouped-query attention.2 None of this is novel, and that is the point. The architecture is deliberately unremarkable so the genomic adaptations sit entirely in tokenization, data, and loss.
The concrete configuration: 30 layers, hidden size 3072, FFN dimension 8448, 32 attention heads with GQA using 4 key-value groups, head dimension 96, RoPE base theta of 5,000,000, and a native context of 32,768 tokens.12 Input and output token embeddings are tied, which regularizes the large DNA sub-vocabulary whose tokens appear infrequently relative to BPE text tokens.2 Carbon-8B shares its layer count, hidden size, feed-forward dimension, and attention configuration exactly with Llama 3-8B.2
Two components carry most of the inference-efficiency story, so they are worth grounding.
Grouped-query attention. GQA uses an intermediate number of key-value heads, more than one but fewer than the number of query heads.6 The motivation is that autoregressive decoder inference is a severe bottleneck "due to the memory bandwidth overhead from loading decoder weights and all attention keys and values at every decoding step."6 Multi-query attention collapses to a single key-value head, which speeds up decoding but can degrade quality. Uptrained GQA recovers near multi-head quality at multi-query speed.6 Carbon-3B's 4 key-value groups against 32 query heads is exactly that compromise, and it shrinks the KV cache that dominates long-context decode.
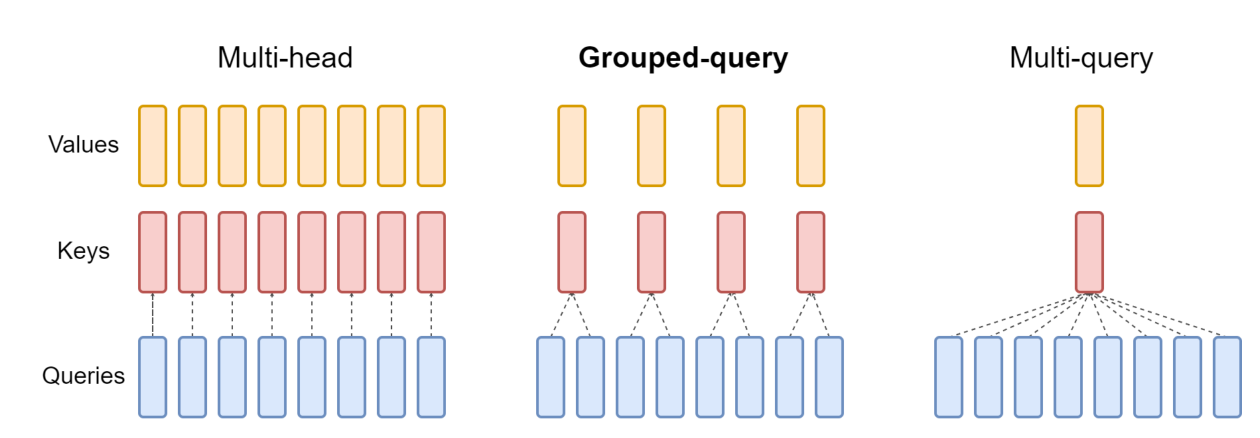
Figure 2: Grouped-query attention sits between multi-head attention (one key-value head per query head) and multi-query attention (a single shared key-value head). Carbon-3B uses 4 key-value groups for 32 query heads. Source: Ainslie et al., "GQA: Training Generalized Multi-Query Transformer Models from Multi-Head Checkpoints" (arXiv:2305.13245).6
Rotary position embeddings. RoPE encodes absolute position with a rotation matrix and folds explicit relative-position dependency directly into the self-attention formulation.7 It gives flexibility in sequence length and a decaying inter-token dependency with increasing relative distance.7 It is also what makes context extension cheap later, because YaRN operates directly on the rotary frequencies. Carbon uses a RoPE base theta of 500,000 (from Llama 3) during base pretraining, then rescales to theta of 5,000,000 in the long-context extension, which is what makes the later YaRN extrapolation tractable.2
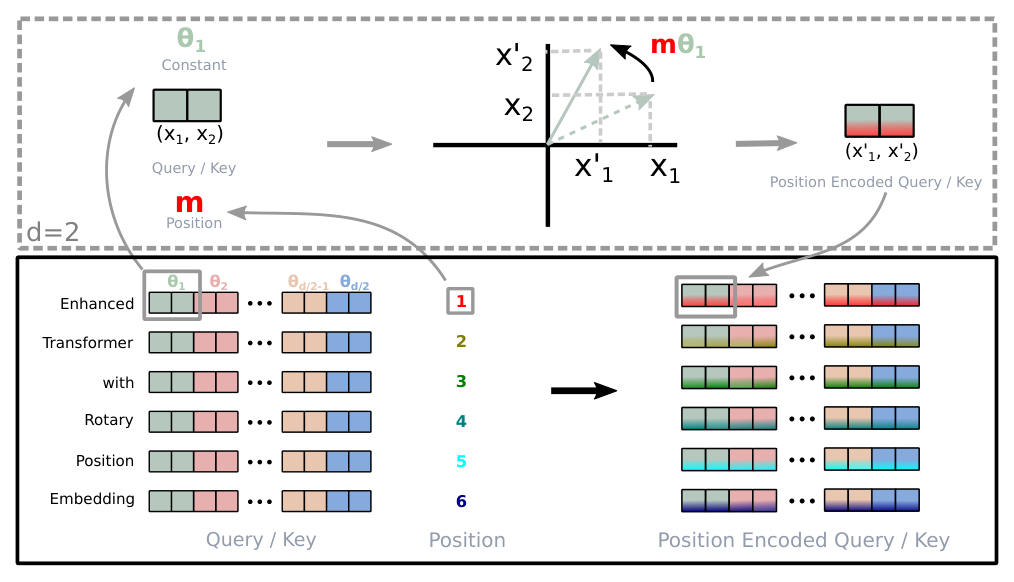
Figure 3: RoPE encodes position as rotations applied per embedding dimension, with the rotation angle scaling with sequence position. Rescaling the base frequency is what later allows YaRN to extend context. Source: Su et al., "RoFormer: Enhanced Transformer with Rotary Position Embedding" (arXiv:2104.09864).7
SwiGLU and RMSNorm round out the stack for the usual reasons. The GEGLU and SwiGLU variants produce the best perplexities among the activations studied, improving over ReLU or GELU.8 RMSNorm drops the re-centering step of LayerNorm on the hypothesis that re-centering invariance is dispensable, which makes it computationally simpler and reduces running time by 7 to 64 percent across models.9 None of this is DNA-specific, which is exactly why it is here.
The tokenizer: a fixed 6-mer scheme, not learned BPE
This is the central DNA-modeling design choice in Carbon, and it is where resolution starts living somewhere other than the input layer. The tokenizer is a hybrid: non-overlapping 6-mer tokenization for DNA combined with the Qwen3 BPE vocabulary for English text.1 Each DNA token encodes 6 nucleotides, so one DNA token is about 6 bp, and a sequence of length L produces roughly L/6 tokens from a vocabulary of 4^6 = 4096 blocks.12 Concretely, the build augments the Qwen3 BPE vocabulary with 4,096 fixed 6-mer tokens, three DNA structural tags, and metadata tokens, for a total vocabulary of 155,776.2
The efficiency argument is direct. Prefill attention cost scales quadratically with token length, so cutting the token count by a factor of six can reduce attention cost for the same nucleotide span by up to a factor of 36.2 Under an 8k-token window, the tokenizer lets Carbon process roughly 8,192 x 6 = 49,152 base pairs.2 This is the mechanism behind the throughput gap in Figure 1.
The interesting decision is not 6-mer versus single-nucleotide. It is fixed 6-mer versus learned BPE, and the justification is not biological mysticism. The authors are explicit: "We choose 6-mer tokenization for Carbon not because DNA is biologically organized in units of six nucleotides. Rather, 6-mer is a simple, deterministic, and largely neutral compression scheme."2 BPE on DNA does the opposite of neutral. It learns frequency-driven merges over a corpus that is half repeats, so the vocabulary ends up encoding transposon boundaries and homopolymer runs rather than anything that transfers to held-out variant tasks. A fixed scheme has nothing to overfit.
The ablation is decisive at matched compute. At 3B parameters and 50B tokens, fixed 6-mer "roughly doubles SR over BPE-32k and BPE-50k (43.25 vs 21.68 / 23.48) and improves BRCA2 AUROC by approximately 10 pp (75.04 vs 63.40 / 66.51) at matched compute."2 That is why the hybrid setup keeps 6-mer for DNA while preserving a BPE vocabulary for future joint English and DNA training.1
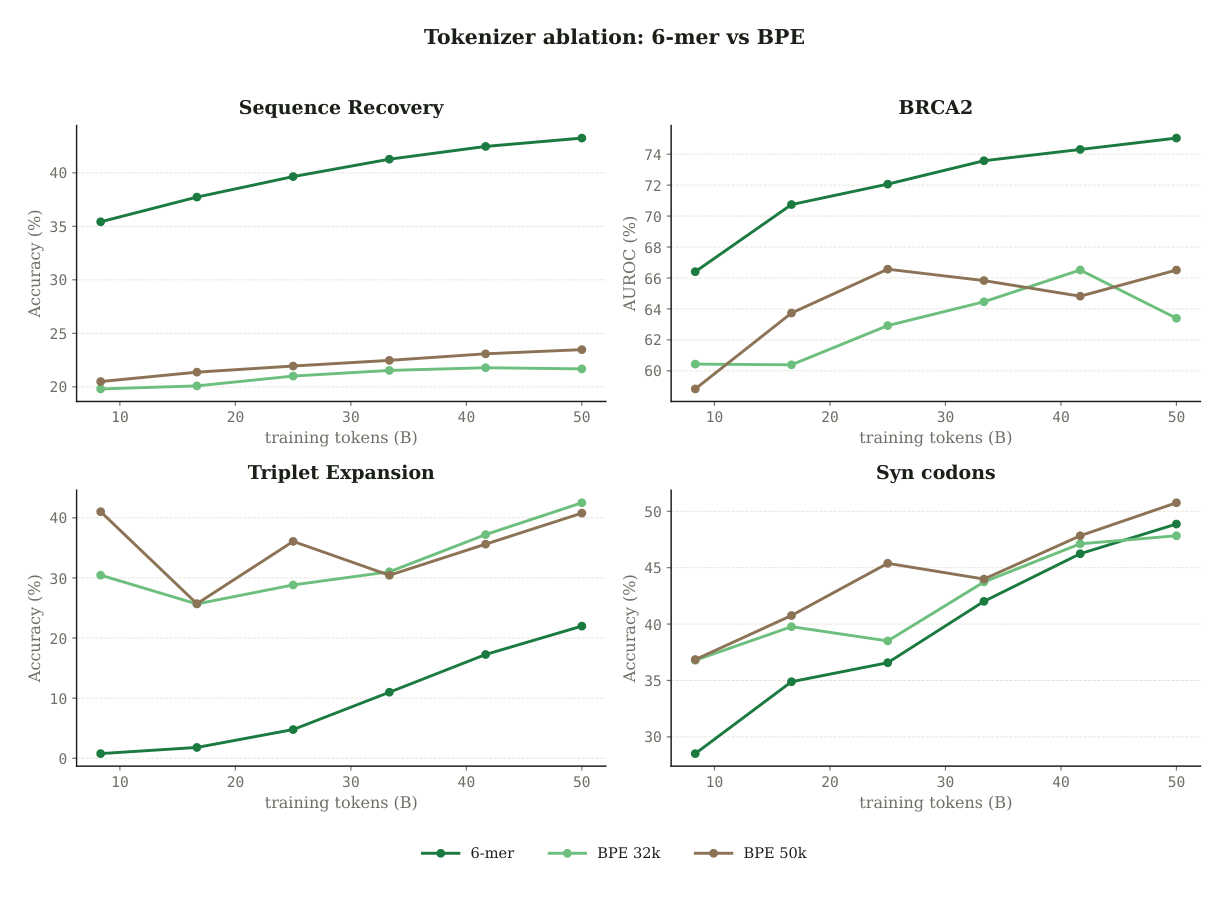
Figure 4: Fixed 6-mer tokenization versus learned BPE at matched compute. The 6-mer scheme dominates sequence recovery and BRCA2 variant scoring. Source: Carbon Technical Report (Hugging Face Biology, 2025).2
The hybrid design has an operational sharp edge. The tokenizer only switches into 6-mer mode when it sees the <dna> tag. Pass a raw DNA string without it, and "the tokenizer treats it as English text and applies BPE. BPE-tokenized DNA is essentially a different language for Carbon-3B, and performance collapses across every benchmark."1 Two more constraints follow: only uppercase A, C, G, T live in the 6-mer vocabulary (lowercase and IUPAC codes map to <oov>), and because each token is exactly 6 bp, input length should be a multiple of 6 or the tokenizer right-pads the trailing block with As.1 Filter to canonical uppercase ACGT and truncate to a multiple of 6 before scoring.
The data: annotation-aware curation over raw nucleotides
If half the genome is repeats and only a couple of percent codes, training on raw sequence wastes most of the budget. Carbon's data strategy is built around the effective density of biological signal rather than raw nucleotide count.2 The arithmetic is stark: "if only 5% of a raw corpus is highly informative, retaining 80% of informative regions while retaining 5% of background regions increases the effective informative fraction from 5% to approximately 46%."2 Same model, same compute, roughly an order of magnitude more signal per step.
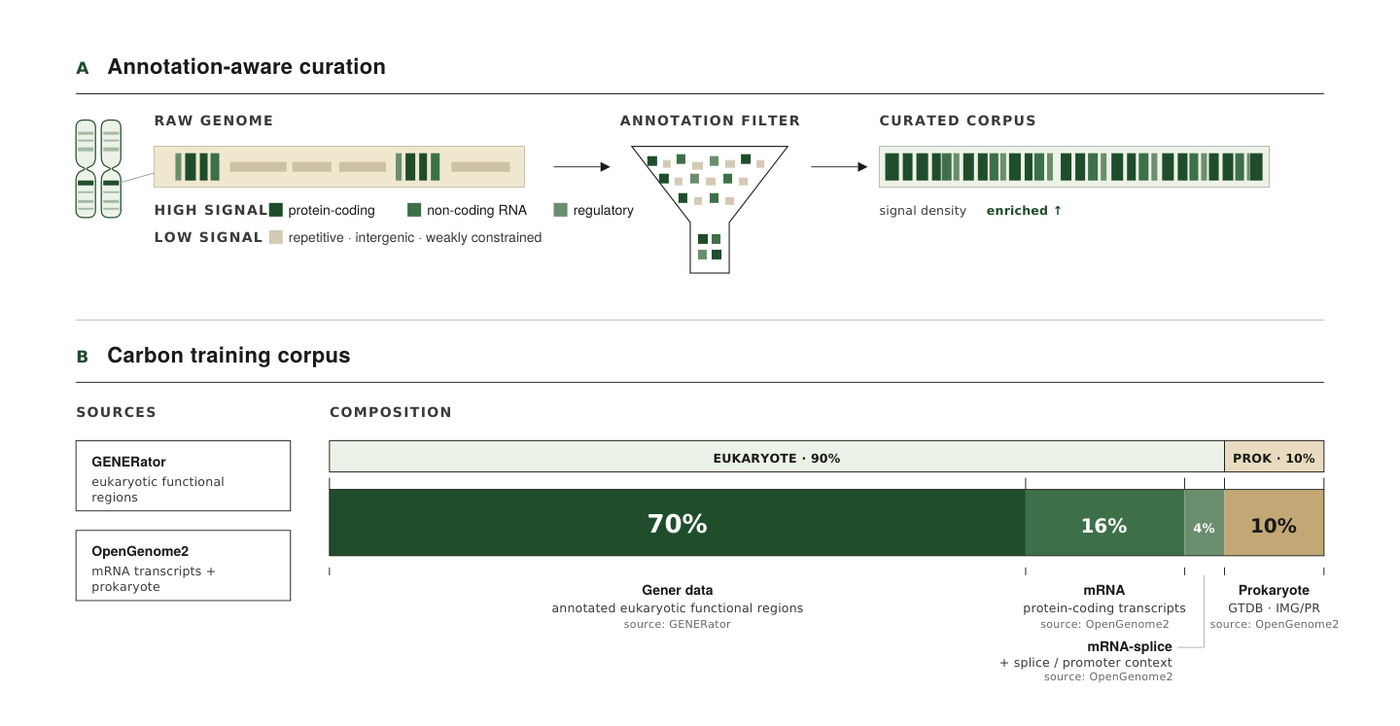
Figure 5: Carbon's annotation-aware curation pipeline and final training mixture. The 90 percent eukaryotic share shown here is the 70 percent Gener genomic-DNA backbone plus the 20 percent eukaryote-derived transcript data. Source: Carbon Technical Report (Hugging Face Biology, 2025).2
The eukaryotic backbone is the GENERator-v2 pretraining corpus, which the report shortens to Gener data; at matched compute it beat OpenGenome2 eukaryote windows on every metric.2 Two further decisions earned their place by ablation. Following GENERator, sequences longer than 100 kbp are dropped, which boosts downstream performance.2 The effect is large: at the 100 kbp filter the model reaches SR 46.38 and BRCA2 81.16 versus 42.75 and 69.05 with no filter, and triplet-expansion accuracy drops from 47.9 percent to 20.5 percent without it.2 Each training sequence is also sampled into one of four prompt templates, some carrying species and gene-type metadata; supplying a species tag improves sequence recovery on the lowest-resource eukaryote groups while leaving high-resource performance unchanged.2
The final mixture that performed best on downstream evaluations is 70 percent eukaryote, 20 percent transcript-level (16 percent mRNA, 4 percent mRNA-splice), and 10 percent prokaryote.2 The prokaryote share is deliberate: adding up to 20 percent prokaryote data leaves eukaryote performance unchanged while improving prokaryote sequence recovery, so the team adopted 10 percent and left the door open for prokaryote continual pretraining later.210 One striking consequence is that Carbon-3B, despite its eukaryotic focus, already matches GENERator-v2-prokaryote-3B, a model trained specifically on prokaryotes, on the prokaryote sequence-recovery split.1
The full corpus is public: 173M DNA and RNA sequences and 1.1 trillion nucleotides, about 180B tokens under the 6-mer tokenizer.11 That number matters for reading the training budget below. The 1T-token pretraining run is therefore roughly six passes over the 180B-token corpus, oversampling the higher-signal configs rather than a single epoch over fresh data.
The training recipe: cross-entropy first, then Factorised Nucleotide Supervision
Here is where resolution moves into the loss, and where Carbon's most distinctive idea lives. When combined with non-overlapping 6-mer tokenization, cross-entropy treats each 6-mer as an atomic class among 4096 possibilities.2 Supervision applies only at the full-token level and does not reflect partial nucleotide agreement.2 A predicted 6-mer that gets five of six bases right is scored exactly like one that gets all six wrong. For variant-effect work, that is the wrong granularity.
Worse, pure cross-entropy training was unstable. In preliminary CE-only runs, the authors observed a recurring instability they call the loss staircase, accompanied by a widening gap between BF16 and FP32 sequence-recovery accuracy. After the staircase, FP32 generation could remain stable or keep improving while BF16 generation degraded substantially.2
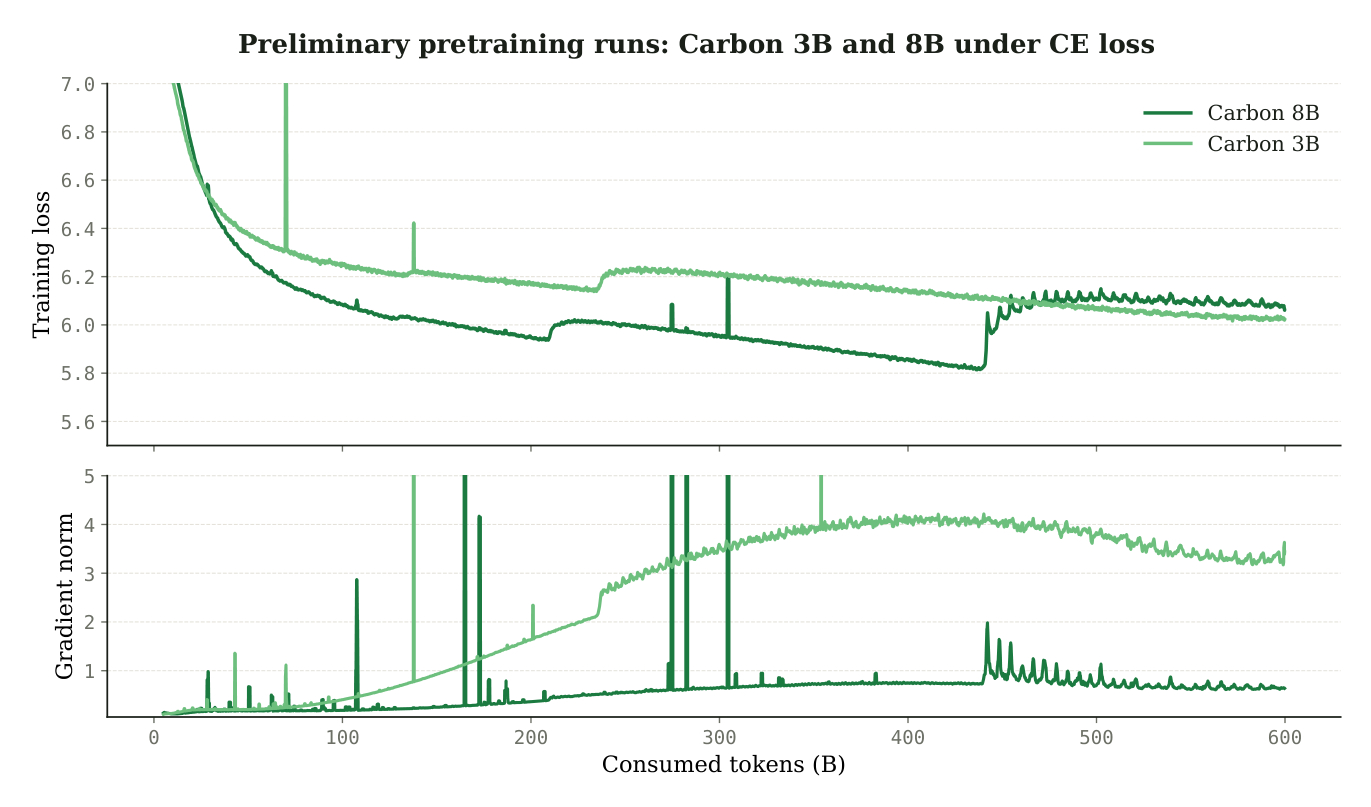
Figure 6: Training dynamics under pure cross-entropy for Carbon-3B and Carbon-8B over the first 600B tokens. The staircase instability and BF16/FP32 divergence motivated the move to a staged objective. Source: Carbon Technical Report (Hugging Face Biology, 2025).2
The answer is Factorised Nucleotide Supervision (FNS). FNS preserves the 6-mer output space but computes the loss through nucleotide-level marginals induced by the 6-mer distribution.2 This gives structured partial credit to near-miss tokens: a 6-mer that matches five positions contributes probability mass to five correct nucleotide marginals, rather than being treated identically to a completely mismatched token.2 Resolution lives in the loss, exactly as promised. The same factorization enables single-nucleotide variant scoring without touching the architecture: marginalizing the 4096-way token distribution into a 4-way nucleotide distribution at the variant position lets a 6-mer model score variants with no change to the tokenizer.2 That trick reappears in the benchmarks and the usage code below.
The team does not train with FNS from scratch. FNS-from-scratch removes the loss staircase but underperforms the staged approach, so the recipe splits the objectives by division of labor: cross-entropy first learns the coarse 6-mer distribution, then FNS sharpens it to nucleotide resolution.2 The schedule for Carbon-3B runs in two phases over 1T 6-mer tokens, about 6T DNA base pairs, at sequence length 8,192 with a global batch of 256 sequences (roughly 2M tokens per step), using AdamW throughout.1
- Phase 1, Cross-Entropy (0 to 100B tokens). Plain CE on the 6-mer vocabulary with a Warmup-Stable-Decay schedule, warmed up over 2,000 iterations to a peak learning rate of 3e-4, then held stable through the end of the phase.12
- Phase 2, Factorised Nucleotide Supervision (100B to 1T tokens). Switch to the hybrid FNS loss at a lower peak learning rate of 5e-5, continuing with a WSD schedule whose decay covers the last 20 percent of Phase-2 steps.2
One detail is worth flagging because the public sources disagree. The technical report states the FNS objective resumes at a peak learning rate of 5e-5, the value used above, while the Carbon-3B model card lists 2e-5.212 Both are small relative to the 3e-4 CE peak and the staging logic is identical either way, but the exact value is ambiguous across the two primary documents. Throughout both phases the loss is computed only on 6-mer DNA token positions; the <dna> and </dna> structural tags are masked and contribute to neither CE nor FNS.2
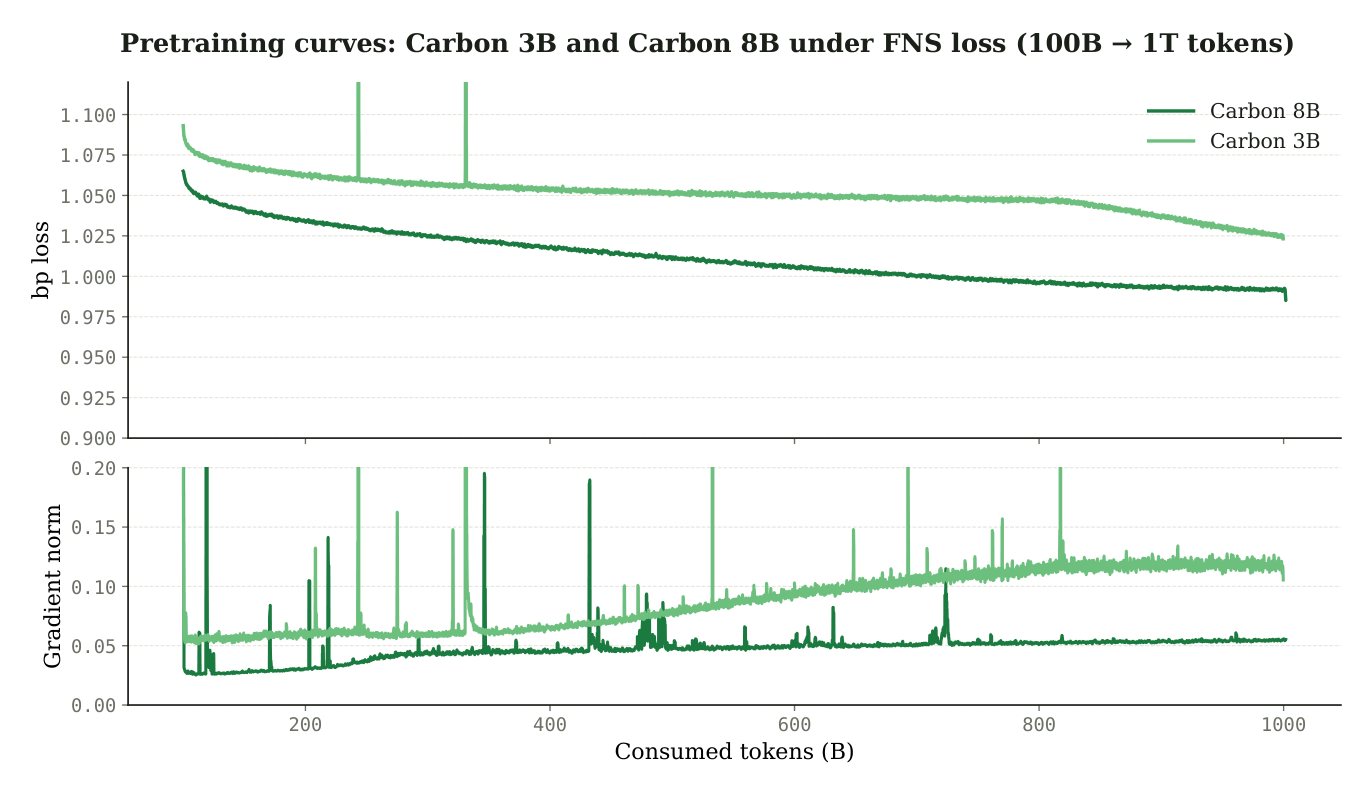
Figure 7: FNS-phase pretraining loss after the CE-to-FNS switch at 100B tokens. The staged schedule descends stably where pure CE did not. Source: Carbon Technical Report (Hugging Face Biology, 2025).2
After base pretraining, the model gets continued training for 50B additional tokens at sequence length 32,768, with the rotary base shifted from 5e5 to 5e6.1 That long-context stage uses a 2,000-step linear warmup from 0 to 3e-5, a stable phase, then a 4,000-step linear decay to 3e-6, at a global batch of 64 sequences of 32,768 tokens, and ran in about 35 hours on 128 H100 GPUs using Megatron-LM with the Carbon patch.1 The result is a native context of 32,768 6-mer tokens, about 197 kbp.1
Benchmarks: parity with Evo2-7B, read honestly
Carbon evaluates with a training-free protocol, meaning it scores frozen pretrained models without task-specific parameter updates.2 The suite spans eight tasks across four capability axes: generation (Sequence Recovery), variant-effect prediction (ClinVar coding and non-coding, BRCA2, TraitGym Mendelian), sequence-level perturbation (nucleotide triplet-expansion and synonymous codon replacement), and long-context retrieval (Genomic-NIAH).2 Single-nucleotide scoring on a 6-mer model works through the marginalization trick from the FNS section: collapse the 4096-way token distribution into a 4-way nucleotide distribution at the variant position, which lets 6-mer models score variants without any architecture change.2
The head-to-head numbers below come from the report's own tables, Carbon-3B versus GENERator-v2-3B versus Evo2-7B.
| Task | Carbon-3B | GENERator-v2-3B | Evo2-7B |
|---|---|---|---|
| Sequence Recovery (eukaryote) | 61.54 | 58.56 | 59.86 |
| BRCA2 (AUROC) | 84.63 | 81.93 | 83.52 |
| ClinVar coding, 24 kb (AUROC) | 92.89 | 91.55 | 93.33 |
| ClinVar non-coding, 24 kb (AUROC) | 91.14 | 90.13 | 89.79 |
| TraitGym Mendelian | 33.65 | 27.91 | 37.78 |
| Nucleotide triplet-expansion | 85.20 | 83.06 | 88.43 |
| Synonymous codon | 88.89 | 87.03 | 91.59 |
Table 1: Training-free benchmark results, Carbon-3B vs GENERator-v2-3B vs Evo2-7B. Higher is better on every metric. Source: Carbon-3B model card (Hugging Face Biology, 2025).1
Read this honestly. Carbon-3B leads on sequence recovery, BRCA2, and ClinVar non-coding; it trails Evo2-7B on TraitGym Mendelian, ClinVar coding, triplet-expansion, and synonymous-codon discrimination. The pattern is parity, not dominance, which is exactly the claim: Carbon-3B is competitive with Evo2-7B while being much faster to run.1 GENERator-v2-3B trails Carbon-3B on every reported task.2
The triplet-expansion and synonymous-codon tasks are constructed to probe single-nucleotide and codon-frame sensitivity, and they are where single-nucleotide-native Evo2 keeps a small edge. Triplet-expansion inserts 10 consecutive CAG triplets into the coding sequence, mimicking polyglutamine expansion disorders such as Huntington's disease; the synonymous-codon task replaces every codon with the species' highest-frequency synonym using CoCoPUTs usage tables.102 A model that has learned codon structure should discriminate the perturbed sequence from the original.
One scope note, stated once: these are the report's own head-to-head tables, not independent reproductions of Evo2 or GENERator-v2, so treat the comparison columns as the report's measurements under its own protocol. Scaling to Carbon-8B closes most of the remaining gaps: it improves over Carbon-3B on every benchmark, with eukaryote sequence recovery rising from 61.54 to 64.05 (+2.51) and the largest gain on long-context retrieval.2
Long context: YaRN extension and the Genomic-NIAH benchmark
Native context for Carbon-3B is 32,768 6-mer tokens, about 197 kbp, extendable to 65,536 tokens (about 393 kbp) at inference via YaRN.1 YaRN (Yet another RoPE extensioN) is a compute-efficient method to extend the context window of RoPE-based models, requiring 10x fewer tokens and 2.5x fewer training steps than previous methods.13 It is the natural extension because Carbon already rescaled the RoPE base for long context, and it applies at inference rather than as another training run.
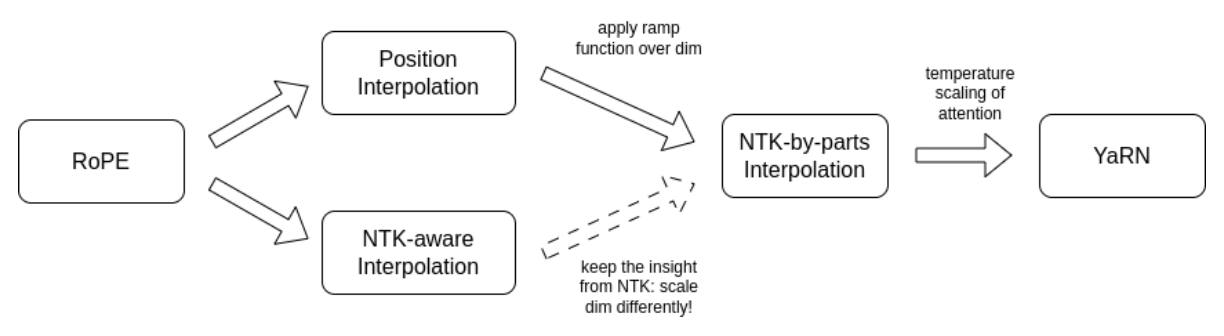
Figure 8: YaRN builds on RoPE interpolation methods, combining NTK-by-parts interpolation with attention temperature scaling. Carbon recommends a YaRN factor of 4. Source: Peng et al., "YaRN: Efficient Context Window Extension of Large Language Models" (arXiv:2309.00071).13
Nominal context length is not the same as usable context length, and Carbon's authors test the difference with a synthetic benchmark. Genomic-NIAH plants a random (KEY, VALUE) DNA pair inside a real-genome haystack and asks the model to recover VALUE given the haystack followed by KEY; the KEY is a random 24 bp DNA string.14 The grid is 4 tasks (plain retrieval plus three near-duplicate variants) crossed with 6 context lengths from 4k to 128k tokens, about 24 kbp to 786 kbp, at 500 examples per cell, for 12,000 retrieval examples total.14 The near-duplicate variants are the diagnostic core: real genomes contain many similar-but-not-identical motifs, so a useful long-context model must discriminate sequences that differ at only a few positions, and the hardest variant places distractor keys that each differ from the target by 4 bp, 83 percent key identity.14 The headline metric is exact-match greedy decoding restricted to DNA tokens, and the haystack is always real genome, never random A/C/G/T padding.14
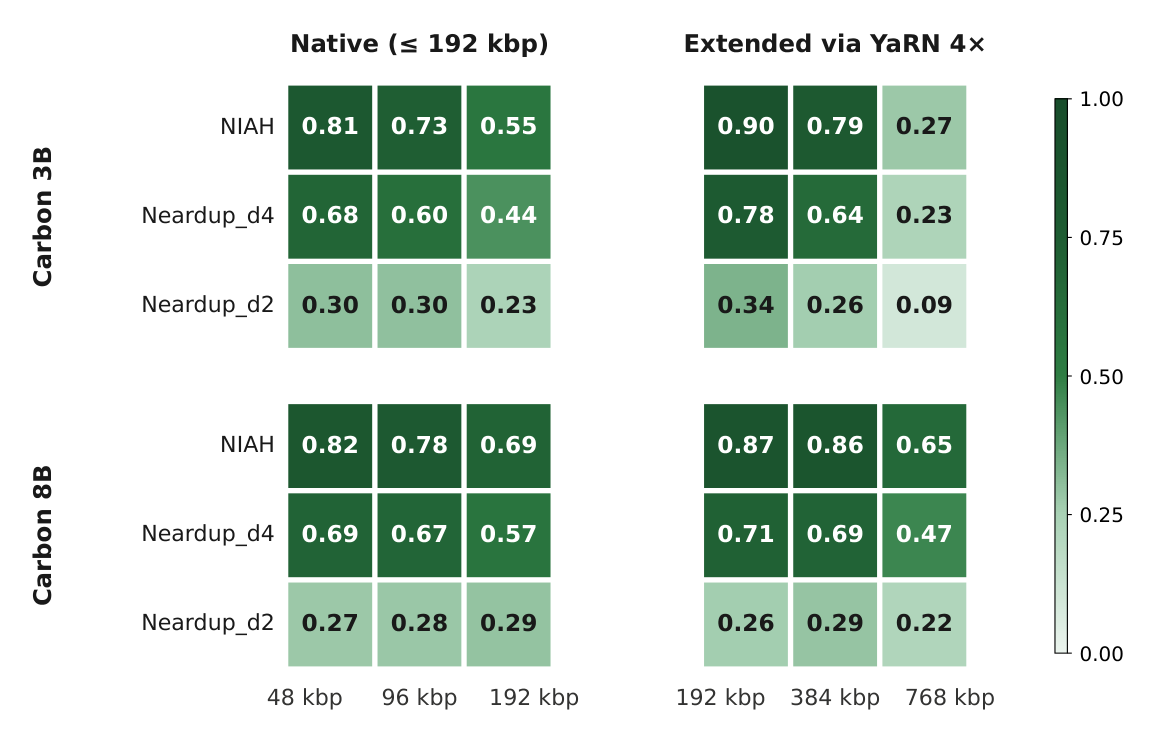
Figure 9: Genomic-NIAH retrieval accuracy across context lengths and difficulty, for Carbon-3B and Carbon-8B, native and YaRN-extended. Source: Carbon Technical Report (Hugging Face Biology, 2025).2
The retrieval results separate nominal from effective context. Carbon-3B retrieves reliably up to its 32k native boundary. YaRN 4x recovers most of the loss at the 32k-to-64k boundary and extends usable retrieval from about 197 kbp to about 384 kbp, then degrades sharply beyond 64k.14 The team recommends a YaRN factor of 4, which gives better retrieval at 64k than a tighter factor of 2, and advises against pushing past 64k tokens, where retrieval collapses at 128k.1 Model size matters: Carbon-3B holds up cleanly at 2x but loses accuracy at 4x, while Carbon-8B extrapolates to 4x without collapse.2
Against Evo2-7B, which was trained natively at 1M tokens, Evo2 leads at short context (16k and 32k) as expected, but Carbon-3B matches it on NIAH at 393 kbp under YaRN despite being substantially smaller.12 At the top end Carbon-8B is ahead of Evo2-7B at both 64k (0.86 vs 0.80) and 128k (0.65 vs 0.53).2 A fair caveat: because Evo2 inference is slow, its NIAH sample sizes are smaller (n=150 at 16k, n=100 at 32k and 64k) than Carbon's n=500 per cell.1
Efficiency: vLLM and speculative decoding
The throughput claim from Figure 1 has a concrete basis. Carbon models run natively in vLLM and generate DNA sequences over 150 times faster than the Evo2 family.1 On a single H100, Carbon-3B generates over 100,000 base pairs per second.1 The speed is structural, not a trick: 6-mer tokenization removes a factor of six from the token count, GQA shrinks the KV cache, and the stock Llama-style backbone slots directly into existing inference engines. The technical report frames the same advantage more conservatively for the apples-to-apples case: in sequence generation and likelihood-based evaluation, Carbon achieves tens-fold faster inference than Evo2 baselines under comparable settings, while requiring several-fold less GPU memory.2
A second efficiency lever is speculative decoding, which the Carbon family was designed to support. The technique samples from autoregressive models faster without changing outputs by computing several tokens in parallel, with a small draft model proposing tokens that the target validates.15 Carbon-500M exists primarily as that draft model: it shares the tokenizer and DNA template format of Carbon-3B and Carbon-8B, so it pairs with either as the target.16 The guarantee is exact: output is identical to greedy decoding with the target alone, and only wall-clock latency drops.16

Figure 10: Speculative decoding accepts multiple draft tokens per target-model step, with rejections corrected by the target model. Carbon-500M serves as the draft model for Carbon-3B and Carbon-8B. Source: Leviathan et al., "Fast Inference from Transformers via Speculative Decoding" (arXiv:2211.17192).15
Carbon-500M is itself a 500M-parameter Llama-style DNA model trained on 600B 6-mer tokens, with the CE-to-FNS switch happening later, at 300B tokens, because its training was very stable, and no long-context stage.16 Pairing it with the 3B target is the intended fast-generation path.
How to use it
Carbon-3B loads as a stock LlamaForCausalLM; only the custom DNA tokenizer needs trust_remote_code=True. The one rule that matters, from the tokenizer section, is non-negotiable: wrap DNA in the <dna> tag and keep length a multiple of 6, or the tokenizer falls back to BPE and quality collapses.1
from transformers import AutoModelForCausalLM, AutoTokenizer
import torch
repo = "HuggingFaceBio/Carbon-3B"
tok = AutoTokenizer.from_pretrained(repo, trust_remote_code=True)
model = AutoModelForCausalLM.from_pretrained(repo, dtype=torch.bfloat16).cuda().eval()
def truncate_to_6mer(seq: str) -> str:
"""Trim a DNA string to a length that is a multiple of 6."""
return seq[: (len(seq) // 6) * 6]
dna = "ATGCGCTAGCTACGATCGATCGTAGCTAGCTAG"
# Open <dna> with no closing tag puts the tokenizer in 6-mer continuation mode.
prompt = f"<dna>{truncate_to_6mer(dna)}"
inputs = tok(prompt, return_tensors="pt", add_special_tokens=False).to("cuda")
out = model.generate(**inputs, max_new_tokens=64, do_sample=False)
generated = tok.decode(out[0, inputs.input_ids.shape[1]:], skip_special_tokens=True)
print(generated)
For variant-effect or perturbation work, score sequences by their per-token log-probabilities. A higher likelihood for the reference than the alternate allele flags the alternate as deleterious. Note the tag asymmetry against the generation snippet: scoring wraps the full sequence in both <dna> and </dna> because the sequence is complete, whereas generation opens <dna> alone to signal a continuation.
import torch
import torch.nn.functional as F
@torch.no_grad()
def score(seq: str) -> float:
"""Mean log-prob per DNA token of `seq` (single sequence, no padding)."""
text = f"<dna>{seq}</dna>" # closed tag scores a fixed sequence
ids = tok(text, return_tensors="pt", add_special_tokens=False).input_ids.to(model.device)
logits = model(ids).logits[:, :-1, :]
targets = ids[:, 1:]
logp = F.log_softmax(logits.float(), dim=-1).gather(-1, targets.unsqueeze(-1)).squeeze(-1)
return logp.mean().item()
# Variant effect = likelihood(alt) - likelihood(ref) over a window around the site.
# delta = score(alt_window) - score(ref_window)
To run beyond the native 32k context, the released config.json targets the native window, so override max_position_embeddings and add a YaRN rope_scaling block at factor 4.
from transformers import AutoConfig, AutoModelForCausalLM
import torch
repo = "HuggingFaceBio/Carbon-3B"
config = AutoConfig.from_pretrained(repo, trust_remote_code=True)
config.max_position_embeddings = 65536 # 65,536 tokens, about 393 kbp
config.rope_scaling = {
# Key is "type" on most releases; some recent transformers versions expect
# "rope_type" instead. Match the form to your installed transformers version.
"type": "yarn",
"factor": 4.0, # factor 4 gives better 64k retrieval than factor 2
"original_max_position_embeddings": 32768,
}
model = AutoModelForCausalLM.from_pretrained(
repo, config=config, dtype=torch.bfloat16,
).cuda().eval()
Speculative decoding is a one-line change: load Carbon-500M and pass it as assistant_model to the 3B target's generate(...) call. The output equals greedy Carbon-3B; only latency drops. Generation can also be conditioned on metadata by prepending species and gene-type tokens, for example <vertebrate_mammalian><protein_coding_region><dna>..., with the unconditional <dna>SEQUENCE</dna> form as the default.1 For full reproducible pipelines, the repo ships seven zero-shot evaluations with canonical scoring scripts.10
Carbon-3B also fine-tunes well on regulatory regression. Fine-tuned on Malinois MPRA, it reaches a mean Pearson correlation of 0.82 across the three cell lines (K562, HepG2, SK-N-SH) on the held-out chromosome 7/13 test set (n=62,582 variants), with the correlation computed separately per cell line.2 That is held-out regression evidence on top of the training-free benchmarks, and the kind of downstream task most groups will actually fine-tune for.
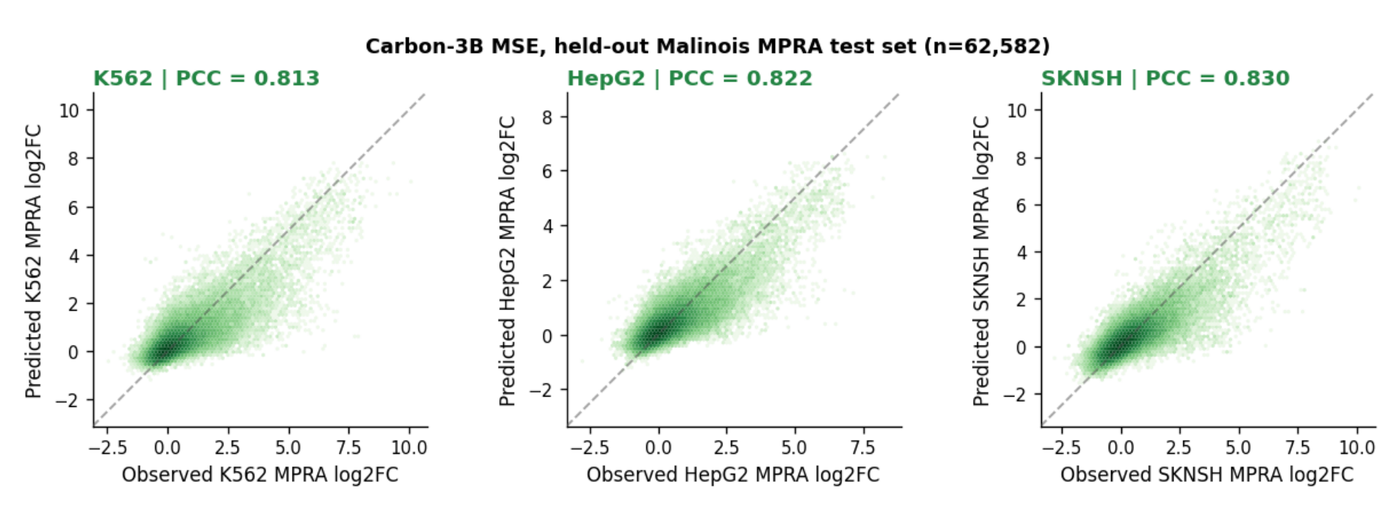
Figure 11: Observed versus predicted log2 fold-change for Carbon-3B fine-tuned on Malinois MPRA across three cell lines (n=62,582 test variants). Source: Carbon Technical Report (Hugging Face Biology, 2025).2
What the model learned, and what it cannot do
The representation analysis is the most direct evidence that the staged 6-mer recipe did not throw away nucleotide-level structure. The team probes two kinds of embeddings and finds a clean division of labor. Separator-token embeddings primarily capture genome-level and taxonomic structure, while content-token embeddings are dominated by local sequence state including strand orientation and codon phase.2 Taxonomic organization sharpens with context: at 8k nucleotides the six taxonomic groups form partially overlapping clouds, and at 48k they resolve into compact, distinct regions.2
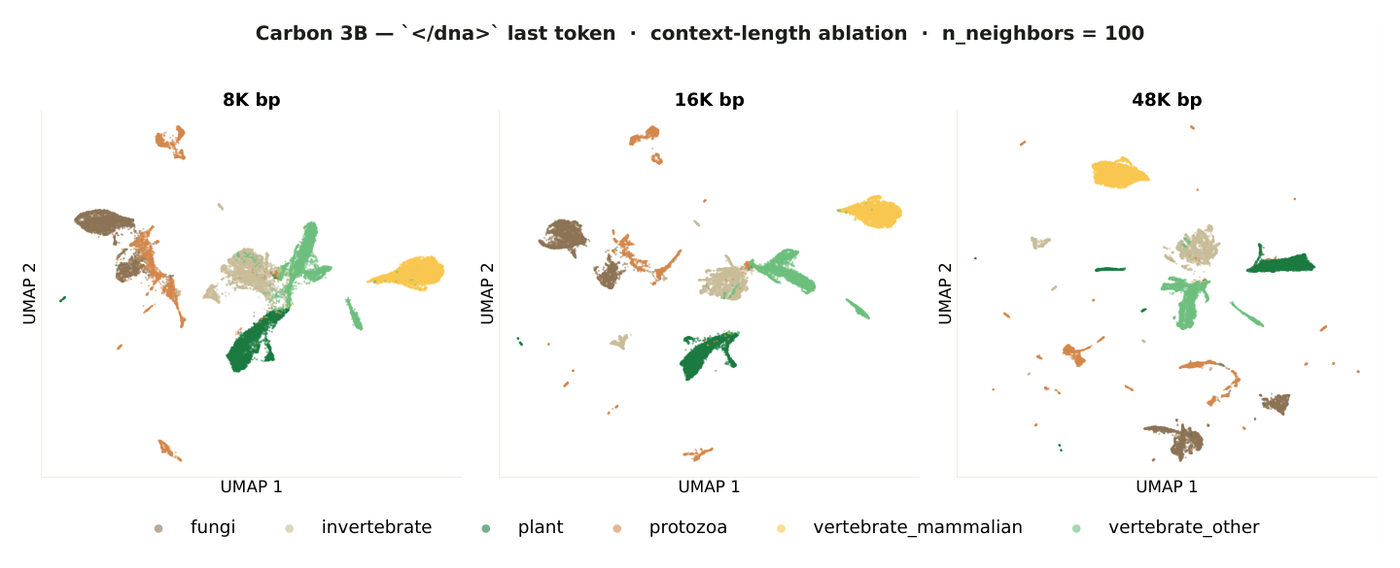
Figure 12: Separator-token embeddings organize by taxonomy as context grows, from overlapping clouds at 8k bp to distinct regions at 48k bp. Source: Carbon Technical Report (Hugging Face Biology, 2025).2
More tellingly, the content-token geometry recovers the natural three-phase structure of coding sequences, which means 6-mer tokenization does not prevent the model from representing reading-frame information.2 That is the resolution-versus-context tension resolved empirically: the loss put resolution back where the tokenizer removed it.
The limitations are real and the authors state them. FNS recovers nucleotide-level supervision from 6-mer logits but relies on a conditional-independence approximation among positions within each token, which improves late-stage stability but is not always biologically correct.2 The two-phase schedule is a workaround for the loss staircase, not a first-principles objective. The context ceiling is honest too: do not push Carbon-3B past 64k tokens, where retrieval collapses, and 393 kbp, while larger than most gene spans, does not cover the longest regulatory domains.1 Carbon-8B's 786 kbp pushes further, but the same architecture does not solve the problem, it scales it.
The broader lesson the authors draw is the one worth keeping: "A model does not need to rely only on the longest nominal context length or the largest parameter count to achieve strong performance if the training pipeline is well aligned with the statistical and biological structure of DNA."2 Put differently: "DNA modeling benefits greatly from LLM technology, but it is not simply natural language modeling with a four-letter alphabet."2
Where to start
The models, the 1.1-trillion-nucleotide corpus, the Genomic-NIAH benchmark, and the full evaluation suite are released.2 Start with the model card at huggingface.co/HuggingFaceBio/Carbon-3B, pull the corpus from carbon-pretraining-corpus, benchmark your own task against genomic-niah, and clone the seven zero-shot evaluations from github.com/huggingface/carbon.
If you have been deferring genome-scale generation or whole-panel variant scoring because Evo2-class inference was too slow, this is the model to re-run that decision against. The open question is not whether 3B is enough, but how much of Carbon's parity comes from the 6-mer tokenizer versus the curation versus the staged objective. The ablations point at all three; disentangling them is the next experiment. Everything starts by wrapping your sequences in <dna> tags. The rest is downstream of that one decision.
References
Footnotes
-
Carbon-3B Model Card. Hugging Face Biology, 2025. https://huggingface.co/HuggingFaceBio/Carbon-3B ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20 ↩21 ↩22 ↩23 ↩24 ↩25 ↩26
-
Ben Allal, L., Li, Q., Fiusco, M., Tunstall, L., Rasul, K., et al. "Carbon: Decoding the Language of Life." Technical Report, Hugging Face Biology, Zhongguancun Academy, TIGEM, University of Naples Federico II, 2025. https://github.com/huggingface/carbon ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8 ↩9 ↩10 ↩11 ↩12 ↩13 ↩14 ↩15 ↩16 ↩17 ↩18 ↩19 ↩20 ↩21 ↩22 ↩23 ↩24 ↩25 ↩26 ↩27 ↩28 ↩29 ↩30 ↩31 ↩32 ↩33 ↩34 ↩35 ↩36 ↩37 ↩38 ↩39 ↩40 ↩41 ↩42 ↩43 ↩44 ↩45 ↩46 ↩47 ↩48 ↩49 ↩50 ↩51 ↩52 ↩53 ↩54 ↩55 ↩56 ↩57 ↩58 ↩59 ↩60 ↩61 ↩62 ↩63 ↩64 ↩65 ↩66 ↩67 ↩68 ↩69
-
Arc Institute. "Evo2: DNA Foundation Model." Preprint, bioRxiv, 2024. https://www.biorxiv.org/content/10.1101/2024.10.22.619039v1 (Evo2 comparison figures here are drawn from the Carbon Technical Report's own head-to-head tables under its evaluation protocol.) ↩
-
Nguyen, E., Poli, M., et al. "HyenaDNA: Long-Range Genomic Sequence Modeling at Single Nucleotide Resolution." NeurIPS, arXiv:2306.15794, 2023. https://arxiv.org/abs/2306.15794 ↩
-
Schiff, Y., Kao, C.-H., Gokaslan, A., et al. "Caduceus: Bidirectional Equivariant Long-Range DNA Sequence Modeling." arXiv:2403.03234, 2024. https://arxiv.org/abs/2403.03234 ↩
-
Ainslie, J., Lee-Thorp, J., de Jong, M., Zemlyanskiy, Y., Lebrón, F., Sanghai, S. "GQA: Training Generalized Multi-Query Transformer Models from Multi-Head Checkpoints." arXiv:2305.13245, 2023. https://arxiv.org/abs/2305.13245 ↩ ↩2 ↩3 ↩4
-
Su, J., Lu, Y., Pan, S., Murtadha, A., Wen, B., Liu, Y. "RoFormer: Enhanced Transformer with Rotary Position Embedding." arXiv:2104.09864, 2021. https://arxiv.org/abs/2104.09864 ↩ ↩2 ↩3
-
Shazeer, N. "GLU Variants Improve Transformer." arXiv:2002.05202, 2020. https://arxiv.org/abs/2002.05202 ↩
-
Zhang, B., Sennrich, R. "Root Mean Square Layer Normalization." arXiv:1910.07467, 2019. https://arxiv.org/abs/1910.07467 ↩
-
Carbon GitHub Repository (README). Hugging Face Biology, 2025. https://github.com/huggingface/carbon ↩ ↩2 ↩3
-
Carbon Pretraining Corpus Dataset Card. Hugging Face Biology, 2025. https://huggingface.co/datasets/HuggingFaceBio/carbon-pretraining-corpus ↩
-
Carbon-3B Model Card (Phase 2 schedule). Hugging Face Biology, 2025. Lists the FNS-phase peak learning rate as 2e-5, which disagrees with the technical report's 5e-5; the report value is used in the main text. https://huggingface.co/HuggingFaceBio/Carbon-3B ↩
-
Peng, B., Quesnelle, J., Fan, H., Shippole, E. "YaRN: Efficient Context Window Extension of Large Language Models." arXiv:2309.00071, 2023. https://arxiv.org/abs/2309.00071 ↩ ↩2
-
Genomic-NIAH Benchmark Dataset Card. Hugging Face Biology, 2025. https://huggingface.co/datasets/HuggingFaceBio/genomic-niah ↩ ↩2 ↩3 ↩4 ↩5
-
Leviathan, Y., Kalman, M., Matias, Y. "Fast Inference from Transformers via Speculative Decoding." arXiv:2211.17192, 2022. https://arxiv.org/abs/2211.17192 ↩ ↩2
-
Carbon-500M Model Card. Hugging Face Biology, 2025. https://huggingface.co/HuggingFaceBio/Carbon-500M ↩ ↩2 ↩3
Comments and feedback
Spotted an error or have a counterpoint? Comment below. No account needed, a name is enough. Corrections and pushback are welcome.