We sequenced the human genome over two decades ago. We can read all three billion letters. But here is the uncomfortable truth: more than 98% of observed genetic variation in humans is non-coding 1, and until last week, we had no single tool that could predict what most of it actually does.
The problem was not a lack of models. It was a surplus. One model for splicing. Another for chromatin accessibility. A third for 3D genome contacts. A fourth for transcription factor binding. Each operated at a different resolution, on a different window of DNA, producing outputs that could not be compared. A researcher trying to understand a single disease-linked variant might need to query half a dozen tools and stitch together their contradictory predictions by hand.
On January 28, 2026, Google DeepMind published AlphaGenome in Nature 2. It takes 1 million base pairs of DNA as input and predicts thousands of functional genomic tracks at single-base-pair resolution across 11 modalities 3. Trained on human and mouse genomes, it matched or exceeded the best available specialized models in 25 of 26 variant effect prediction evaluations 4. The patchwork era may be ending.
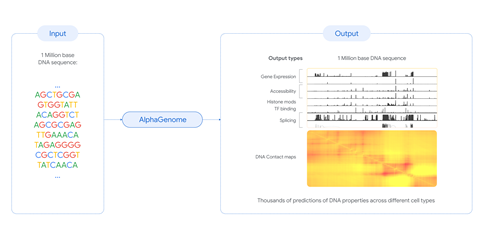 AlphaGenome reads raw DNA and simultaneously predicts gene expression, chromatin accessibility, histone modifications, transcription factor binding, splicing, and 3D contact maps across cell types. Source: Chemistry World
AlphaGenome reads raw DNA and simultaneously predicts gene expression, chromatin accessibility, histone modifications, transcription factor binding, splicing, and 3D contact maps across cell types. Source: Chemistry World
Why This Was Hard
Building a unified genomic model required breaking two trade-offs that had constrained the field for years.
The first was resolution versus range. Models like SpliceAI and BPNet provided base-pair-resolution predictions but were "restricted to short input sequences (for example, 10 kb or less), and thus may miss the influence of distal regulatory elements" 5. An enhancer 500,000 base pairs upstream? Invisible. Models like Enformer and Borzoi extended the context window to 200-500 kb "to capture broader context but at the cost of reducing output resolution (128-bp or 32-bp bins)" 6. Fine-scale features like splice sites and individual transcription factor footprints got blurred into coarse averages.
The second was breadth versus depth. SpliceAI predicts splicing. ChromBPNet predicts chromatin accessibility. Orca predicts 3D chromatin contacts. Each excelled at its task but offered zero insight into the others. As Ziga Avsec of DeepMind put it: "Previously, the field required separate models for separate tasks. AlphaGenome unifies these under one roof" 7.
Why had nobody unified them sooner? Processing a million base pairs at single-nucleotide resolution is computationally brutal. The memory and compute costs scale with sequence length, and doing it across 11 modalities simultaneously multiplies the challenge. Enformer, the previous generation, took twice the compute budget to train on sequences five times shorter 8. The engineering problem was real, and AlphaGenome solves it through a combination of architectural efficiency and sequence parallelism.
Architecture: CNNs Read Words, Transformers Read Paragraphs
AlphaGenome uses "a U-Net-inspired backbone architecture" that produces representations at three scales: 1-bp, 128-bp, and 2,048-bp resolution 9. If you have worked with image segmentation models, the structure is familiar: an encoder downsamples the input, a decoder upsamples it back, and skip connections preserve fine-grained detail at each level.
The design splits two complementary jobs between two types of layers. "Convolutional layers model local sequence patterns necessary for fine-grained predictions, whereas transformer blocks model coarser but longer-range dependencies in the sequence, such as enhancer-promoter interactions" 10. The convolutions read individual words in the genome; the transformers understand how sentences relate to each other across a very long document.
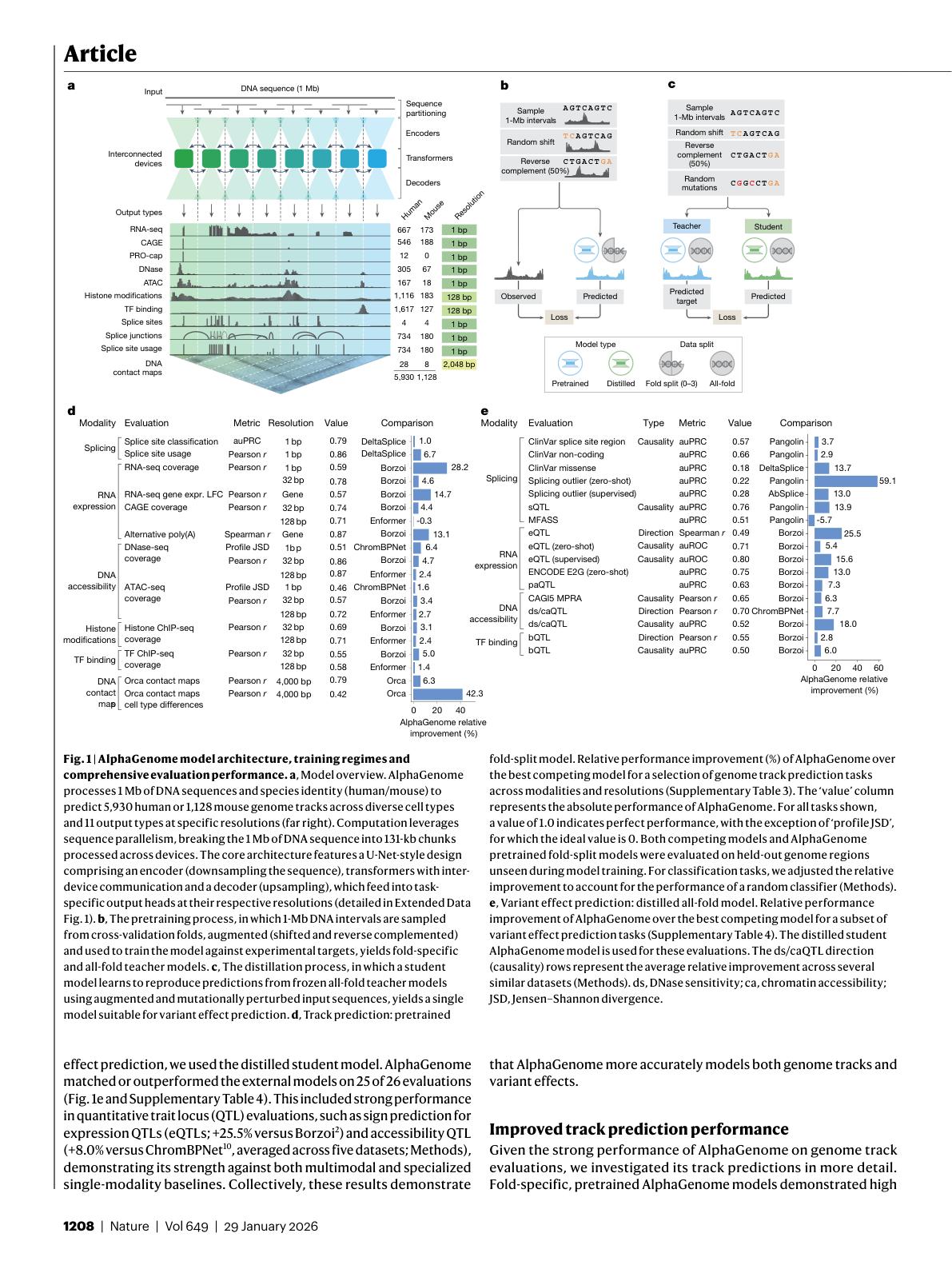 Figure 1: AlphaGenome architecture showing the U-Net encoder-decoder design, pre-training with cross-validation, distillation for variant scoring, and benchmark performance. Source: Benegas et al., Nature (2026), Figure 1
Figure 1: AlphaGenome architecture showing the U-Net encoder-decoder design, pre-training with cross-validation, distillation for variant scoring, and benchmark performance. Source: Benegas et al., Nature (2026), Figure 1
"Base-pair-resolution training on the full 1-Mb sequence is enabled through sequence parallelism across eight interconnected tensor processing unit (v3) devices" 11. The 1 Mb sequence gets broken into 131 kb chunks processed in parallel 12. Training follows two stages: first, pretraining with 4-fold cross-validation on data from ENCODE, GTEx, 4D Nucleome, and FANTOM5 13 14; then distillation, where a student model learns from ensemble teacher predictions while also processing mutated input sequences, producing a compact model optimized for variant scoring.
The efficiency numbers are striking. "Training a single AlphaGenome model (without distillation) took four hours and required half of the compute budget used to train our original Enformer model" 8. The distilled student model runs inference in under one second on an NVIDIA H100 GPU 15. More capability with less compute is the kind of engineering advance that compounds over time.
The 1 Mb context window was chosen deliberately: "99% (465 of 471) of validated enhancer-gene pairs fall within 1 Mb" 16. The model sees nearly all known long-range regulatory relationships for any given gene.
Beating 25 of 26 Specialized Models
AlphaGenome "simultaneously predicts 5,930 human or 1,128 mouse genome tracks across 11 modalities" 17. The benchmark results are unusually clean. For single-sequence predictions, "AlphaGenome outperformed these external models on 22 of 24 evaluations" 18. For variant effect prediction, it "matched or outperformed the external models on 25 of 26 evaluations" 19.
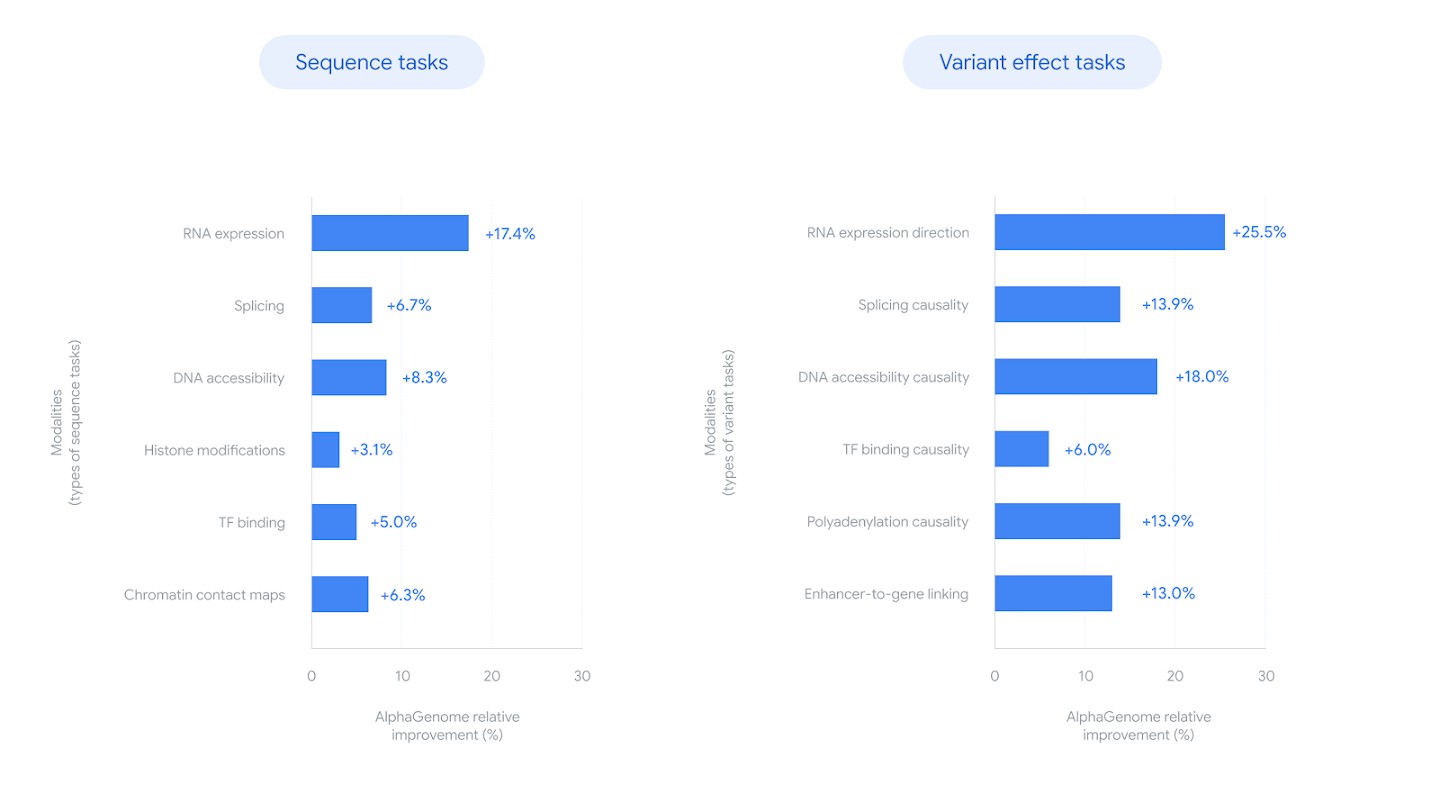 Relative improvement of AlphaGenome over the best competing model across sequence prediction tasks (left) and variant effect prediction tasks (right). Source: Google DeepMind Blog
Relative improvement of AlphaGenome over the best competing model across sequence prediction tasks (left) and variant effect prediction tasks (right). Source: Google DeepMind Blog
The most important gains:
- Gene expression: +14.7% relative improvement in cell-type-specific gene-level expression prediction compared with Borzoi 20. For eQTL sign prediction, +25.5% versus Borzoi 21.
- eQTL recovery: At a threshold yielding 90% sign prediction accuracy, "AlphaGenome recovered over twice as many GTEx eQTLs (41%) as Borzoi (19%)" 22. This is the number that matters most for practical variant interpretation: twice as many disease-relevant variants correctly classified.
- Contact maps: +6.3% over Orca on contact map Pearson correlation, and +42.3% for cell-type-specific differences 23.
- Polyadenylation: Spearman rho of 0.894 versus Borzoi's 0.790 24.
David Kelley of Calico Life Sciences, who led development of both Enformer and Borzoi, acknowledged the advance: "I think the long sequence length that they're able to work with here is definitely one of those major engineering breakthroughs" 25.
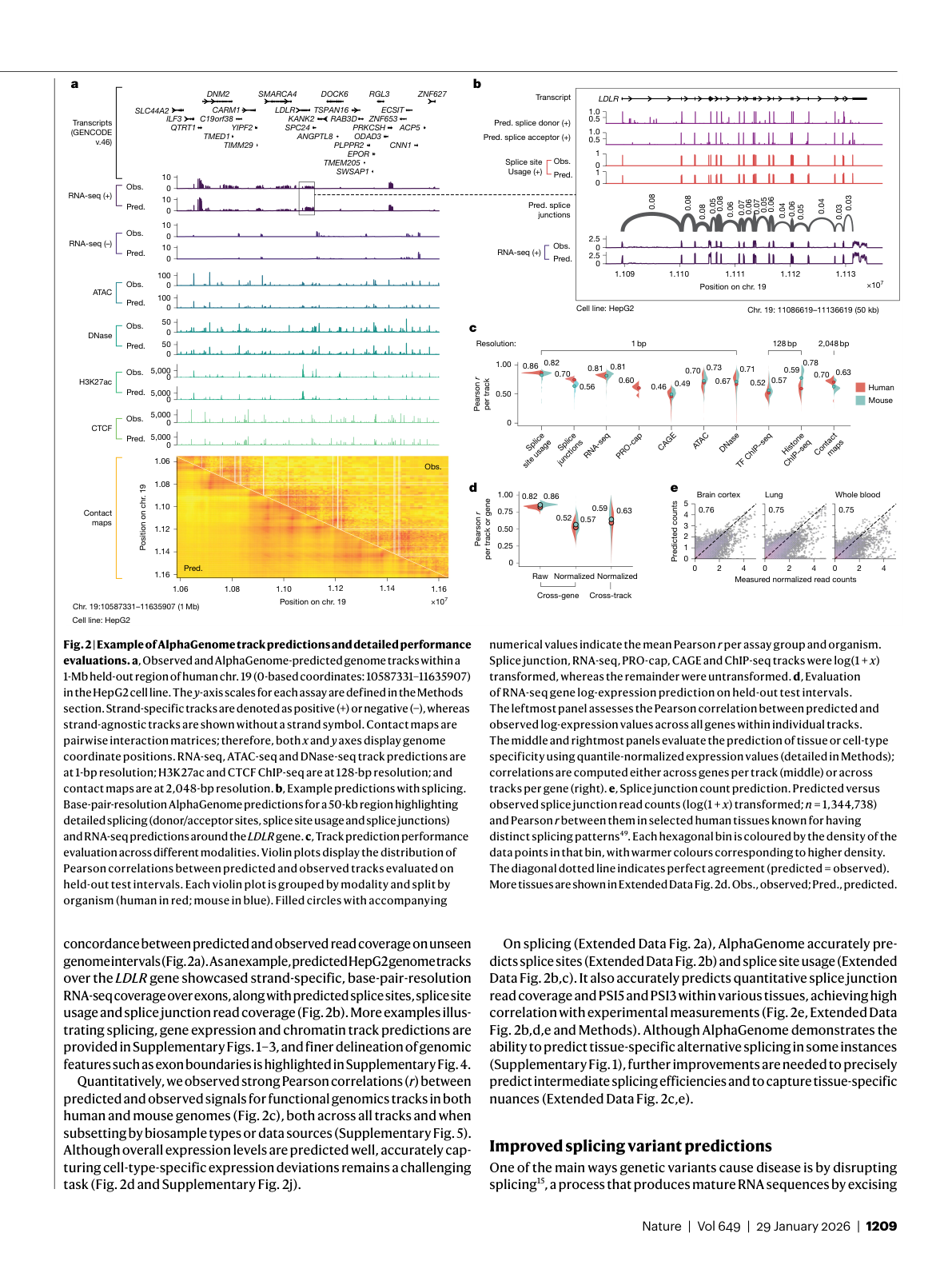 Figure 2: Observed versus predicted genome tracks in the HepG2 cell line, including RNA-seq, ATAC-seq, histone marks, and chromatin contact maps. Source: Benegas et al., Nature (2026), Figure 2
Figure 2: Observed versus predicted genome tracks in the HepG2 cell line, including RNA-seq, ATAC-seq, histone marks, and chromatin contact maps. Source: Benegas et al., Nature (2026), Figure 2
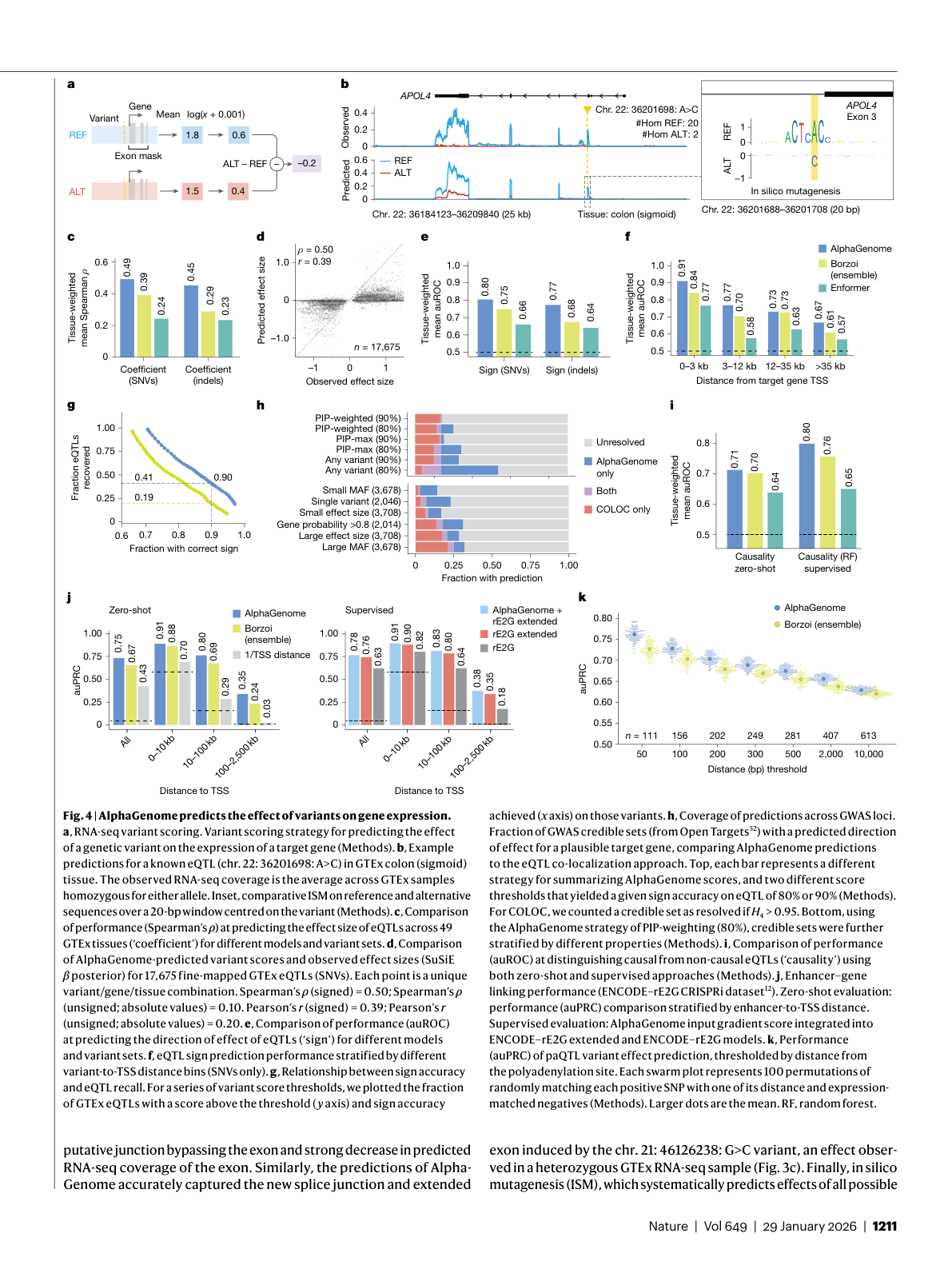 Figure 4: RNA-seq variant scoring strategy, eQTL predictions, GWAS loci coverage, and enhancer-gene linking benchmarks. Source: Benegas et al., Nature (2026), Figure 4
Figure 4: RNA-seq variant scoring strategy, eQTL predictions, GWAS loci coverage, and enhancer-gene linking benchmarks. Source: Benegas et al., Nature (2026), Figure 4
The multimodal architecture is not just a convenience feature. Ablation studies showed that "the fully multimodal model generally outperformed models trained on single modality groups, confirming the overall benefit of integrating diverse data types for learning shared representations" 26. Training on everything together teaches the model cross-modal relationships that single-task models cannot learn. Models trained on longer sequences also outperformed those trained on shorter sequences, even when both were evaluated with 1 Mb context 27, suggesting that training-time exposure to long-range patterns matters independently of inference-time context.
Splicing: Where It Gets Clinical
Splicing prediction is where AlphaGenome's clinical relevance becomes most concrete. "It is estimated that overall, as many as 1 in 3 disease-associated single-nucleotide variants are splice-disruptive" 28. Diseases like spinal muscular atrophy and some forms of cystic fibrosis "can be caused by errors in RNA splicing" 29.
Previous models like SpliceAI could predict whether a splice site exists, but not how it competes with neighboring splice sites for usage, or how a variant might create an entirely new junction. AlphaGenome predicts all three quantities simultaneously, providing "a more comprehensive view of the splicing-related molecular consequences of variants" 30. On ClinVar pathogenic variant classification, "AlphaGenome composite scores outperformed the existing best method in each category across all three variant categories: deep intronic and synonymous (auPRC 0.66 versus 0.64 by Pangolin), splice region (auPRC 0.57 versus 0.55 by Pangolin) and missense (auPRC 0.18 versus 0.16 by DeltaSplice and Pangolin)" 31.
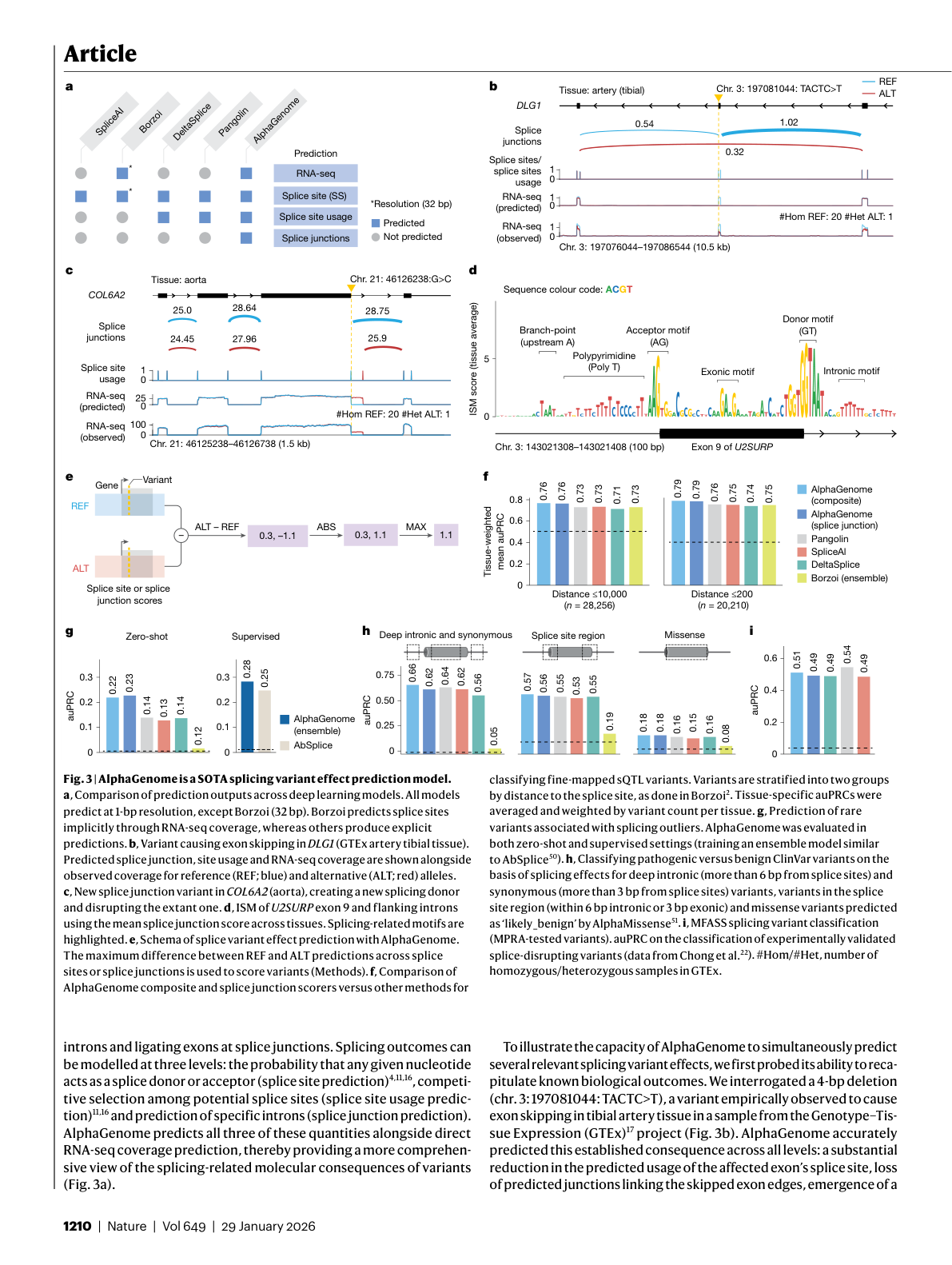 Figure 3: Splicing variant effect prediction performance across benchmarks, comparing AlphaGenome with SpliceAI, Pangolin, and other specialized models. Source: Benegas et al., Nature (2026), Figure 3
Figure 3: Splicing variant effect prediction performance across benchmarks, comparing AlphaGenome with SpliceAI, Pangolin, and other specialized models. Source: Benegas et al., Nature (2026), Figure 3
Cancer Variant Interpretation: Connecting the Dots
The real power of a unified model shows up when you need to trace a variant's effects across multiple molecular layers. The paper demonstrates this through a case study of known cancer-associated mutations near the TAL1 oncogene, a gene whose inappropriate activation drives T-cell acute lymphoblastic leukemia (T-ALL), a form of childhood leukemia.
DeepMind "used AlphaGenome to investigate the potential mechanism of a cancer-associated mutation" 32. For one oncogenic mutation (chr. 1: 47239296: C>ACG), "AlphaGenome predicted increases in the activating histone marks H3K27ac and H3K4me1 at the variant, consistent with experimentally observed neo-enhancer formation at that position" 33. Histone marks like H3K27ac and H3K4me1 are chemical tags on DNA-packaging proteins that signal active gene regulation. The model predicted that the mutations would activate TAL1 "by introducing a MYB DNA binding motif, which replicated the known disease mechanism" 34.
A splicing-only model would have missed this variant entirely. An expression-only model might have flagged the change but could not have explained the mechanism. AlphaGenome connected the dots across histone modifications, transcription factor binding, and gene expression in a single inference pass.
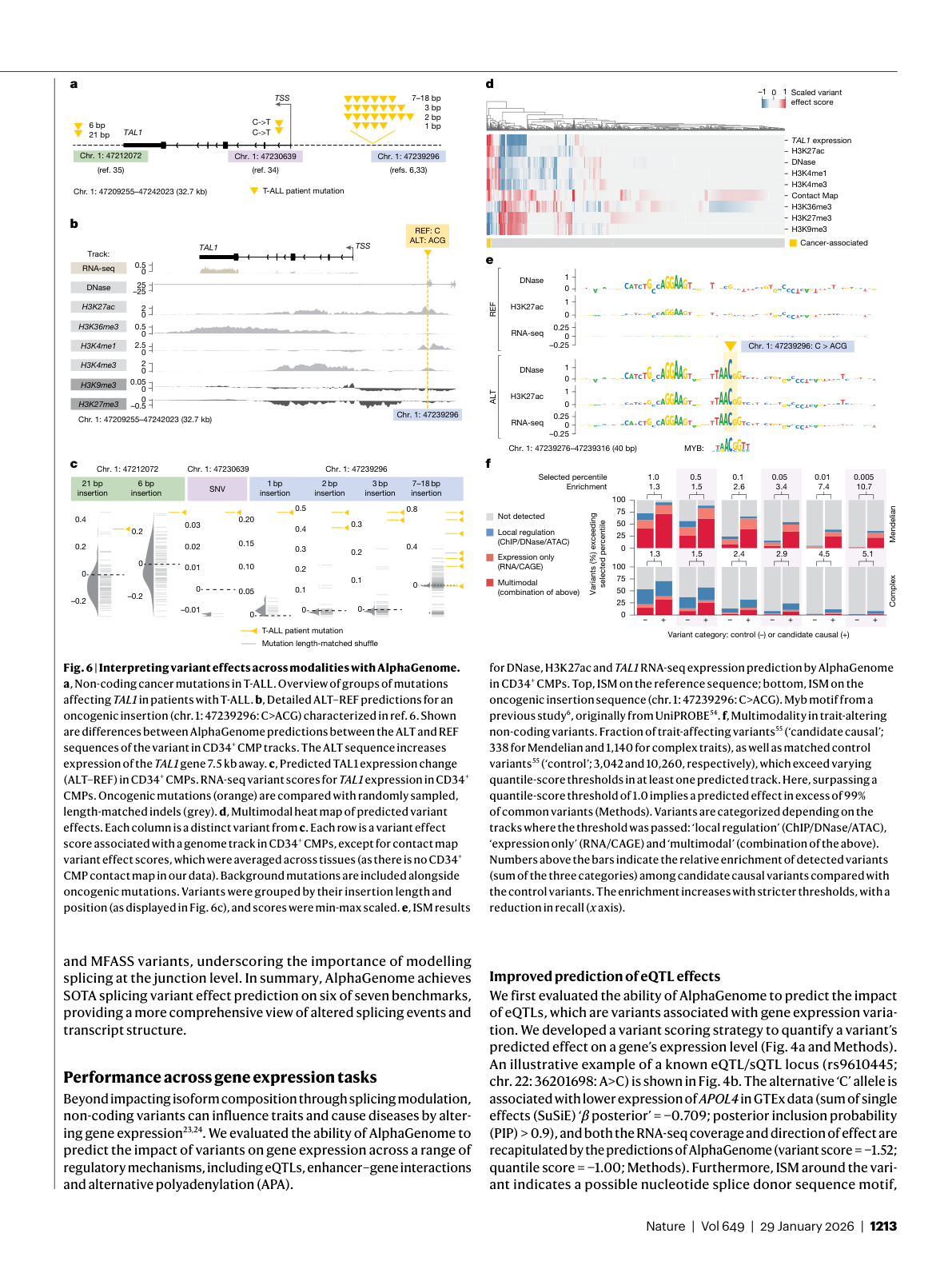 Figure 6: Cross-modal variant interpretation for T-ALL cancer mutations near TAL1. AlphaGenome simultaneously predicts changes in histone marks, transcription factor binding, accessibility, and gene expression. Source: Benegas et al., Nature (2026), Figure 6
Figure 6: Cross-modal variant interpretation for T-ALL cancer mutations near TAL1. AlphaGenome simultaneously predicts changes in histone marks, transcription factor binding, accessibility, and gene expression. Source: Benegas et al., Nature (2026), Figure 6
Professor Marc Mansour of University College London commented: "AlphaGenome will be a powerful tool for the field. Determining the relevance of different non-coding variants can be extremely challenging, particularly to do at scale. This tool will provide a crucial piece of the puzzle, allowing us to make better connections to understand diseases like cancer" 35.
What AlphaGenome Cannot Do
The limitations are important, and the DeepMind team is upfront about them.
Distant regulatory elements remain hard. "Accurately capturing the influence of distal regulatory elements (more than 100 kb away) remains a continuing objective" 36. Even with 1 Mb of context, some regulatory interactions span greater distances.
Species coverage is narrow. "Our species coverage remains limited to human and mouse, and the evaluations in this study are primarily human-focused" 37.
Molecular predictions are not disease predictions. AlphaGenome predicts molecular consequences of variants, but "application to complex trait analysis is limited because AlphaGenome predicts molecular consequences of variants, whereas these phenotypes involve broader biological processes" 38. Ziga Avsec put it plainly: "Predicting how a disease manifests from the genome is an extremely hard problem, and this model is not able to magically predict that" 39. The gap between "this variant changes H3K27ac signal at this locus" and "this patient will develop disease X" remains vast. Clinical-grade variant interpretation still requires integration with population data, functional assays, and expert review through established frameworks like the ACMG/AMP guidelines, the standard clinical system for classifying genetic variants 40.
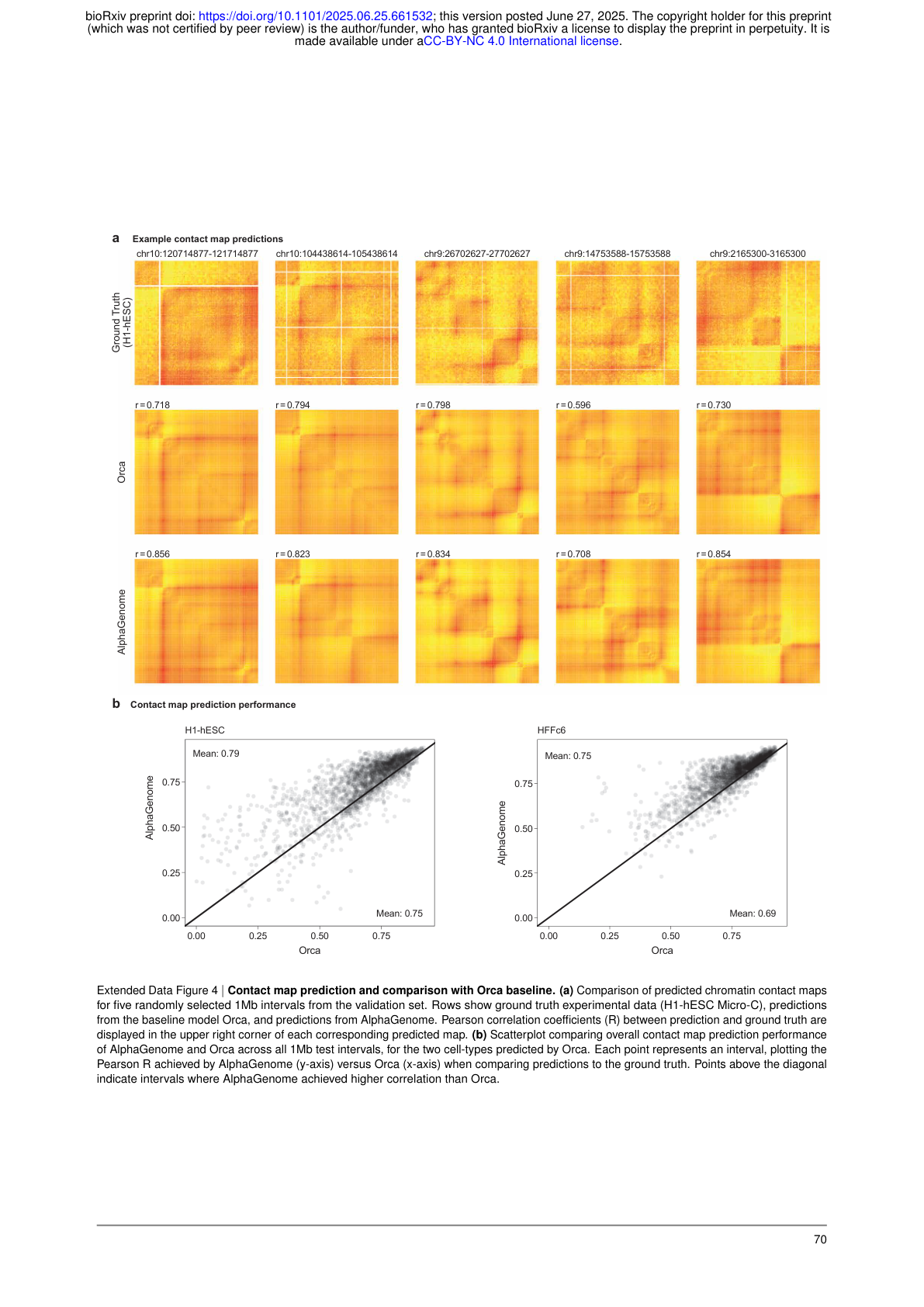 Extended Data Figure 4: AlphaGenome achieves higher Pearson R values than Orca on chromatin contact map predictions across randomly selected 1 Mb intervals. Source: Benegas et al., bioRxiv (2025), Extended Data Fig. 4
Extended Data Figure 4: AlphaGenome achieves higher Pearson R values than Orca on chromatin contact map predictions across randomly selected 1 Mb intervals. Source: Benegas et al., bioRxiv (2025), Extended Data Fig. 4
Interpretability is limited. Jian Zhou of the University of Chicago observed: "Even when the model makes accurate predictions, it does not always directly inform us of the underlying biological processes" 41. The model tells you what will change. It does not always tell you why.
Remaining Questions
AlphaGenome is available for non-commercial use through an online API 42, and adoption has been rapid: nearly 3,000 scientists in 160 countries have used it, submitting around 1 million requests each day 43.
Several open questions remain. Can the multimodal training approach scale to additional species? How will AlphaGenome interact with generative DNA models? The authors note that "AlphaGenome can complement the capabilities of generative models trained on DNA sequences by predicting functional properties of newly generated sequences" 44, a combination that could accelerate synthetic biology. The model "assigned a confident sign prediction for at least one variant in 49% of GWAS credible sets" 45, but getting from molecular prediction to complex trait analysis remains a separate, harder problem.
A broader question lingers. Compared with the well-defined problem of protein structure prediction, "genomics is more of a fuzzy field. There's no single metric of success," as Natasha Latysheva of DeepMind noted 46. AlphaFold had one clear target: match the experimentally determined 3D structure. AlphaGenome must predict thousands of functional measurements across hundreds of cell types. The evaluation surface is vastly more complex.
AlphaGenome predicts what happens to chromatin, expression, and splicing. It does not yet predict what happens to the patient. That gap is the field's next problem, and closing it will take years of experimental validation that no model can shortcut. But for the first time, any researcher with an internet connection can query the regulatory consequences of a DNA variant across all major functional modalities in under a second. What they do with that capability will determine whether AlphaGenome's impact approaches AlphaFold's.
References
Footnotes
-
Ibid. ↩
-
Benegas et al., Nature (2026). Op. cit. ↩
-
Benegas et al., "Advancing regulatory variant effect prediction with AlphaGenome," Nature (2026). https://www.nature.com/articles/s41586-025-10014-0 ↩
-
Ibid. ↩
-
Ibid. ↩
-
Ibid. ↩
-
Ziga Avsec, quoted in Chemistry World (2026). https://www.chemistryworld.com/news/googles-alphagenome-wants-to-do-for-dna-what-alphafold-did-for-proteins/4022824.article ↩
-
Ibid. ↩
-
Ibid. ↩
-
Ibid. ↩
-
Benegas et al., bioRxiv preprint (2025). https://www.biorxiv.org/content/10.1101/2025.06.25.661532v1 ↩
-
Google DeepMind Blog, "AlphaGenome: AI for better understanding the genome" (2026). https://deepmind.google/blog/alphagenome-ai-for-better-understanding-the-genome/ ↩
-
Ibid. ↩
-
Ibid. ↩
-
Ibid. ↩
-
Ibid. ↩
-
Ibid. ↩
-
Ibid. ↩
-
Ibid. ↩
-
Ibid. ↩
-
Ibid. ↩
-
Ibid. ↩
-
Ibid. ↩
-
David Kelley, Calico Life Sciences, quoted in Scientific American (2026). https://www.scientificamerican.com/article/google-deepmind-unleashes-new-ai-alphagenome-to-investigate-dnas-dark-matter/ ↩
-
Ibid. ↩
-
Ibid. ↩
-
Smith & Kitzman, "Benchmarking splice variant prediction algorithms using massively parallel splicing assays," Genome Biology (2023). https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-03144-z ↩
-
Google DeepMind Blog (2026). Op. cit. ↩
-
Benegas et al., Nature (2026). Op. cit. ↩
-
Ibid. ↩
-
Ibid. ↩
-
Ibid. ↩
-
Ibid. ↩
-
Professor Marc Mansour, University College London, quoted in DeepMind Blog (2026). Ibid. ↩
-
Ibid. ↩
-
Ibid. ↩
-
Ibid. ↩
-
Ziga Avsec, quoted in Scientific American (2026). Ibid. ↩
-
Richards et al., "Standards and guidelines for the interpretation of sequence variants," Genetics in Medicine 17:405-424 (2015). https://www.nature.com/articles/gim201530 ↩
-
Jian Zhou, quoted in Chemistry World (2026). Ibid. ↩
-
Ibid. ↩
-
Pushmeet Kohli, quoted in Chemistry World (2026). Ibid. ↩
-
Ibid. ↩
-
Ibid. ↩
-
Natasha Latysheva, quoted in STAT News (2025). https://www.statnews.com/2025/06/25/google-ai-deepmind-launches-alphagenome-new-model-to-predict-dna-encoding-gene-regulation/ ↩