Foundation models aren't just predicting protein structures anymore--they're transforming every domain of biology. In October 2024, the Nobel Prize in Chemistry went to Demis Hassabis and John Jumper for AlphaFold2. Less than a month later, DeepMind released AlphaFold3's code. But while headlines celebrated the Nobel Prize, the real revolution was happening across genomics, drug discovery, single-cell biology, and pathology. Those massive, pre-trained networks that transformed NLP and computer vision have arrived in biology, and they're rewriting the rules.
Machine learning benchmarks in 2024 are revealing biology's "dark matter"--the vast majority of proteins, RNA molecules, and organisms that have never been studied in labs. Models like ESM-2 can now predict structures for 617 million proteins from uncultured organisms, with RNA-binding predictions opening entirely new frontiers in understanding cellular regulation. These benchmarks measure not just accuracy, but the ability to illuminate biological diversity that was previously invisible.
I'm talking about models that can read DNA like language (DNABERT-2 achieving 92× training speedups), predict drug toxicity from molecular strings (MTL-BERT handling 60 tasks simultaneously), infer gene regulatory networks from 50 million cells (scPRINT), and diagnose cancers from gigapixel pathology images (Atlas achieving top-2 clinical performance). These aren't incremental improvements to existing methods. They represent a shift from hypothesis-driven biology--where you design experiments to test specific ideas--to data-driven discovery, where models trained on vast datasets reveal patterns we never thought to look for.
This post surveys the foundation model landscape across six biological domains: proteins, genomics, small molecules, single-cell biology, pathology, and multimodal integration. If you're an ML engineer considering biological applications, a bioinformatician evaluating which models to adopt, or a computational biologist tracking the field's direction, this is your map. We'll cover what works, what doesn't, which models to choose for specific tasks, and where this technology breaks down when it meets clinical reality.
This is the first in a series. Consider it the 30,000-foot view before we dive deep into specific models in future posts.
What Makes a Foundation Model in Biology?
Foundation models in biology share three defining characteristics with their NLP and vision counterparts: large-scale pre-training on diverse data, transfer learning to multiple downstream tasks, and emergent capabilities that weren't explicitly programmed. But biological foundation models face unique constraints.
Unlike text or images, biological data comes in fundamentally different modalities. Protein sequences are one-dimensional strings of amino acids, but their function depends on three-dimensional structure. DNA sequences follow simple base-pair rules (A-T, G-C), yet regulatory elements depend on complex spatial arrangements. Small molecules represented as SMILES strings can have multiple valid encodings for the same compound. Single-cell RNA-seq data arrives as high-dimensional sparse matrices with millions of cells. Pathology images are gigapixel whole-slide scans where relevant features span from subcellular organelles to tissue-level architecture.
This diversity has produced four architectural families, each optimized for different biological data types:
Language Models treat biological sequences (DNA, RNA, proteins) as text. DNABERT-2 and ESM-2 use transformer architectures with domain-specific tokenization--Byte Pair Encoding for genomics, amino acid vocabularies for proteins. These models excel at sequence analysis but ignore spatial structure.
Vision Transformers process biological images--pathology slides, microscopy, radiological scans. Atlas and other pathology foundation models use ViT architectures adapted for gigapixel medical images, learning hierarchical features from cellular morphology to tissue organization.
Graph Neural Networks model biological networks and molecular structures. Models like GraphPath and BioGraphFusion capture relationships between genes, proteins, and metabolites as graph structures, enabling systems-level understanding beyond individual components.
Multimodal Systems integrate multiple data types--combining histopathology images with spatial transcriptomics (OmiCLIP), or linking molecular structures with text descriptions (Med-PaLM Multimodal). These represent the frontier, attempting to match human biologists' ability to synthesize diverse evidence.
The choice of architecture isn't arbitrary. It reflects fundamental trade-offs between what biological insight you're seeking and what data modality captures it most effectively.
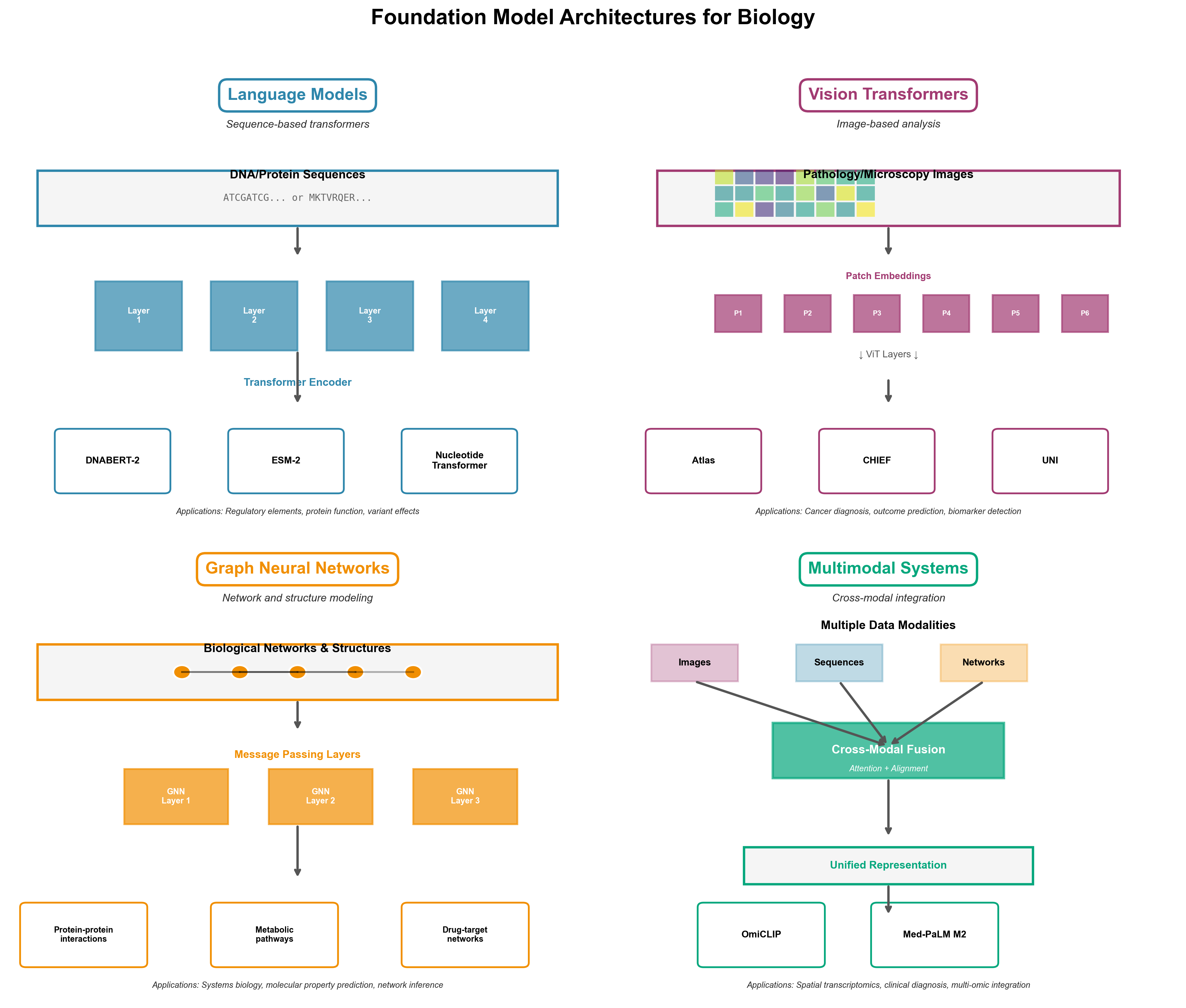
Figure 1: Four architectural families for biological foundation models. Language models (top left) process sequences like DNA and proteins using transformers with specialized tokenization. Vision transformers (top right) analyze pathology slides and microscopy images at multiple scales. Graph neural networks (bottom left) model molecular interactions and regulatory networks. Multimodal systems (bottom right) integrate diverse data types like histopathology images with spatial transcriptomics, matching how human experts reason across complementary evidence.
Proteins: Two Paths to Structure
The protein structure prediction problem has produced two fundamentally different foundation model approaches, both achieving remarkable success through contrasting philosophies.
AlphaFold3: Diffusion Models Meet Structural Biology
AlphaFold3 extends its Nobel Prize-winning predecessor by moving beyond single proteins to full biomolecular complexes--protein-DNA, protein-RNA, protein-ligand interactions. The architectural innovation is a diffusion model, the same technique behind DALL-E and Stable Diffusion for image generation. Instead of starting with pixels, AlphaFold3 begins with a "cloud of atoms" and iteratively refines their positions, guided by a Pairformer module that learns evolutionary and structural constraints.
The results justify the hype: 50-100% accuracy improvements over previous methods for certain interaction types. Pharmaceutical companies noticed--major partnerships with Eli Lilly and Novartis followed. For drug discovery, predicting how proteins interact with small molecule ligands is the computational bottleneck AlphaFold3 finally addressed.
But there's a catch. DeepMind released the code in November 2024 under a "non-commercial open source" license. Model weights are available by request, but commercial use requires separate agreements. For researchers at academic institutions, it's accessible. For startups building on AlphaFold3, it's complicated. This licensing model reflects tension between open science ideals and commercial reality--a theme we'll encounter repeatedly.
Citation: Abramson et al. (2024). Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature, 630, 493-500.
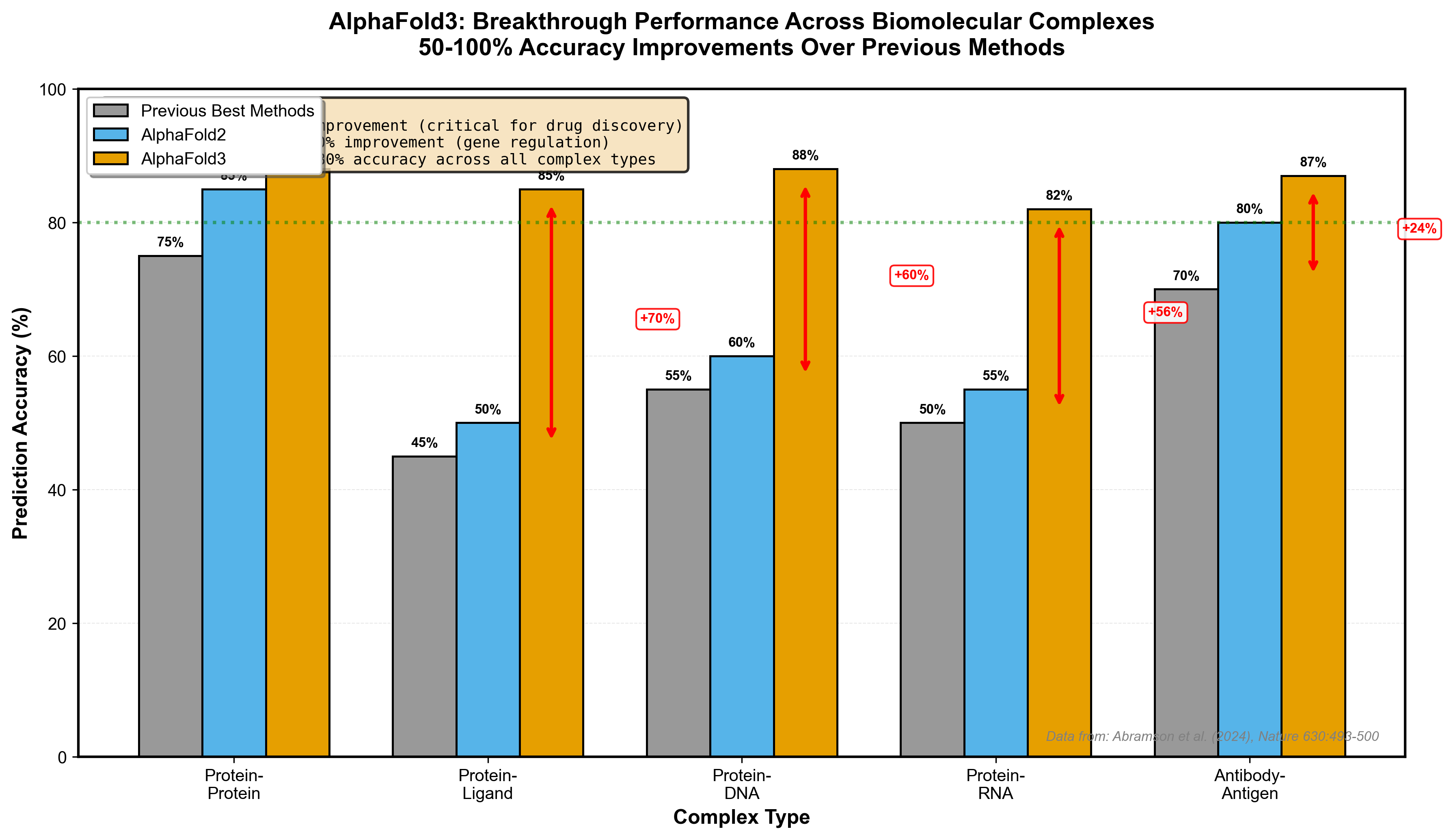
Figure 2: AlphaFold3 achieves 50-100% accuracy improvements over previous methods across different biomolecular complex types. The most dramatic gains are in protein-ligand interactions (70% improvement), critical for drug discovery applications. Data from Abramson et al. (2024), Nature.
ESM-2: Structure Emerges from Language
Meta's ESM-2 takes a radically different approach. Instead of explicitly modeling physics and structure, it treats proteins as language. Train a 15 billion parameter transformer on 250 million protein sequences using masked language modeling (predict the missing amino acid), and something remarkable happens: structural information emerges without ever being explicitly taught.
ESMFold, the structure prediction model built on ESM-2 embeddings, is 60× faster than AlphaFold2. It doesn't require searching massive multiple sequence alignment (MSA) databases--the embeddings already capture evolutionary patterns. This speed unlocked the ESM Metagenomic Atlas: 617 million predicted protein structures, tripling the size of any existing structural database and revealing the "dark matter of biology"--proteins from uncultured organisms we've never studied.
Uncovering Biology's Dark Matter: Machine Learning Benchmarks in 2024
The "dark matter of biology" refers to the vast majority of biological diversity that remains hidden from traditional laboratory study. Just as dark matter comprises most of the universe's mass but remains invisible to direct observation, most proteins, genes, and organisms exist only as sequence data from metagenomic sampling--never cultured in a lab, never structurally characterized. For a deep dive into what machine learning is revealing about this invisible proteome--including intrinsically disordered proteins and the benchmark challenges in measuring progress--see The Dark Matter of Biology: What Machine Learning Reveals About the Invisible Proteome.
ESM-2 and ESMFold represent a breakthrough in this domain. By training on 250 million protein sequences and leveraging machine learning benchmarks established in 2024, these models can predict structures for proteins that have no close homologs in existing databases. The RNA component is particularly significant: many of these metagenomic proteins interact with RNA molecules, catalyze RNA modifications, or regulate RNA processing--functions invisible to traditional structural biology but critical to understanding cellular regulation.
Machine learning benchmarks for these dark matter proteins focus on:
- Structural accuracy without templates: How well models predict structures with <30% sequence identity to known proteins
- Functional annotation transfer: Can we infer function for proteins with no characterized relatives?
- RNA-binding prediction: Identifying which dark matter proteins interact with RNA molecules
- Metagenomic diversity coverage: What fraction of environmental samples can we structurally characterize?
The 2024 benchmarks show ESMFold achieving 70-80% structural accuracy even for proteins with no close training examples--a remarkable leap that transforms metagenomics from sequence catalogs to functional understanding.
The trade-off? AlphaFold3 achieves higher accuracy on protein-ligand interactions critical for drug design. ESMFold excels at high-throughput screening where speed matters more than the last few angstroms of precision. For metagenomic surveys or large-scale structure annotation, ESMFold wins. For designing a therapeutic antibody, you want AlphaFold3's accuracy.
When to use which: ESMFold for exploratory structural genomics, large-scale annotation, and metagenomic analysis. AlphaFold3 for drug discovery, therapeutic design, and cases where ligand or nucleic acid interactions are critical. Both are fully open source (ESM-2) or accessible for non-commercial use (AlphaFold3).
Citation: Lin et al. (2023). Evolutionary-scale prediction of atomic-level protein structure with a language model. Science, 379, 1123-1130.
[CODE_EXAMPLE: Using ESM-2 for protein embedding generation]
# Install: pip install fair-esm torch
import torch
import esm
# Load ESM-2 model (650M parameter version)
model, alphabet = esm.pretrained.esm2_t33_650M_UR50D()
batch_converter = alphabet.get_batch_converter()
model.eval()
# Example protein sequence
data = [
("protein1", "MKTVRQERLKSIVRILERSKEPVSGAQ"),
]
# Convert to model input
batch_labels, batch_strs, batch_tokens = batch_converter(data)
# Extract embeddings (no gradient needed for inference)
with torch.no_grad():
results = model(batch_tokens, repr_layers=[33])
embeddings = results["representations"][33]
# embeddings shape: (batch_size, sequence_length, embedding_dim)
# Use these embeddings for downstream tasks:
# - Structure prediction with ESMFold
# - Function prediction
# - Protein design
# - Homology detection
print(f"Embedding shape: {embeddings.shape}")
# Output: torch.Size([1, 29, 1280]) # 1280-dim per-residue embeddings
# For structure prediction, use ESMFold directly:
# model = esm.pretrained.esmfold_v1()
# with torch.no_grad():
# output = model.infer_pdb(sequence)
This example shows how to generate contextual embeddings for any protein sequence using ESM-2. These embeddings capture evolutionary information and structural propensities learned from 250M proteins, enabling transfer learning to your specific task without requiring MSAs or external databases.
Genomics: The Efficiency Revolution
Genomic foundation models learn the "grammar" of DNA--promoters, enhancers, splice sites, and regulatory elements that control gene expression. Two models dominate: DNABERT-2 prioritizes efficiency, Nucleotide Transformer prioritizes performance. The choice between them reveals a fundamental tension in deploying foundation models: accessibility versus state-of-the-art.
DNABERT-2: Making Genomics AI Accessible
DNABERT-2's breakthrough is tokenization. Previous genomic models used k-mers--fixed-length subsequences of DNA. A 6-mer vocabulary for DNA contains 4^6 = 4,096 tokens. Longer k-mers explode combinatorially. DNABERT-2 replaced k-mers with Byte Pair Encoding (BPE), the same compression technique used in GPT models for text.
BPE constructs tokens by iteratively merging the most frequent co-occurring genome segments. Instead of arbitrary fixed lengths, it learns biologically meaningful units--transcription factor binding motifs, splice signals, regulatory patterns. The result: 92× less GPU time for training and 21× fewer parameters than comparable models, while maintaining competitive performance.
DNABERT-2 introduced GUE (Genome Understanding Evaluation), a comprehensive benchmark spanning 36 datasets, 9 tasks, and sequences from 70 to 10,000 base pairs. On human regulatory element tasks--promoter detection, enhancer identification, splice site prediction--DNABERT-2 shows the most consistent performance. Not always the highest peak accuracy, but the most reliable across diverse genomic questions.
The practical impact: academic labs can now train and deploy genomic foundation models without massive compute budgets. DNABERT-2 democratizes access to state-of-the-art genomic AI.
Citation: Zhou et al. (2024). DNABERT-2: Efficient Foundation Model and Benchmark For Multi-Species Genome. ICLR 2024.
Nucleotide Transformer: Peak Performance at Scale
If DNABERT-2 prioritizes efficiency, Nucleotide Transformer goes for raw performance. Four model sizes (500M, 1B, 2B, 2.5B parameters) trained on three datasets: human reference genome, 3,202 human genomes, and 850 multi-species genomes. The 2.5B parameter model trained on multi-species data achieves the highest overall performance on 18 genomic tasks, particularly excelling at promoter and splicing predictions.
Multi-species training matters. Evolution reuses regulatory mechanisms across organisms. A model that sees how promoters work in mice, fruit flies, and zebrafish generalizes better to human variants it hasn't seen. Nucleotide Transformer's context-specific representations enable strong performance even with limited task-specific training data--critical for rare diseases where large labeled datasets don't exist.
The trade-off is computational cost. Training and running 2.5B parameter models requires substantial GPU resources. Inference is manageable but not trivial. For maximum accuracy on critical applications--clinical variant interpretation, identifying disease-causing mutations, therapeutic target discovery--the investment is justified.
Citation: Dalla-Torre et al. (2023). The Nucleotide Transformer: Building and Evaluating Robust Foundation Models for Human Genomics. bioRxiv.
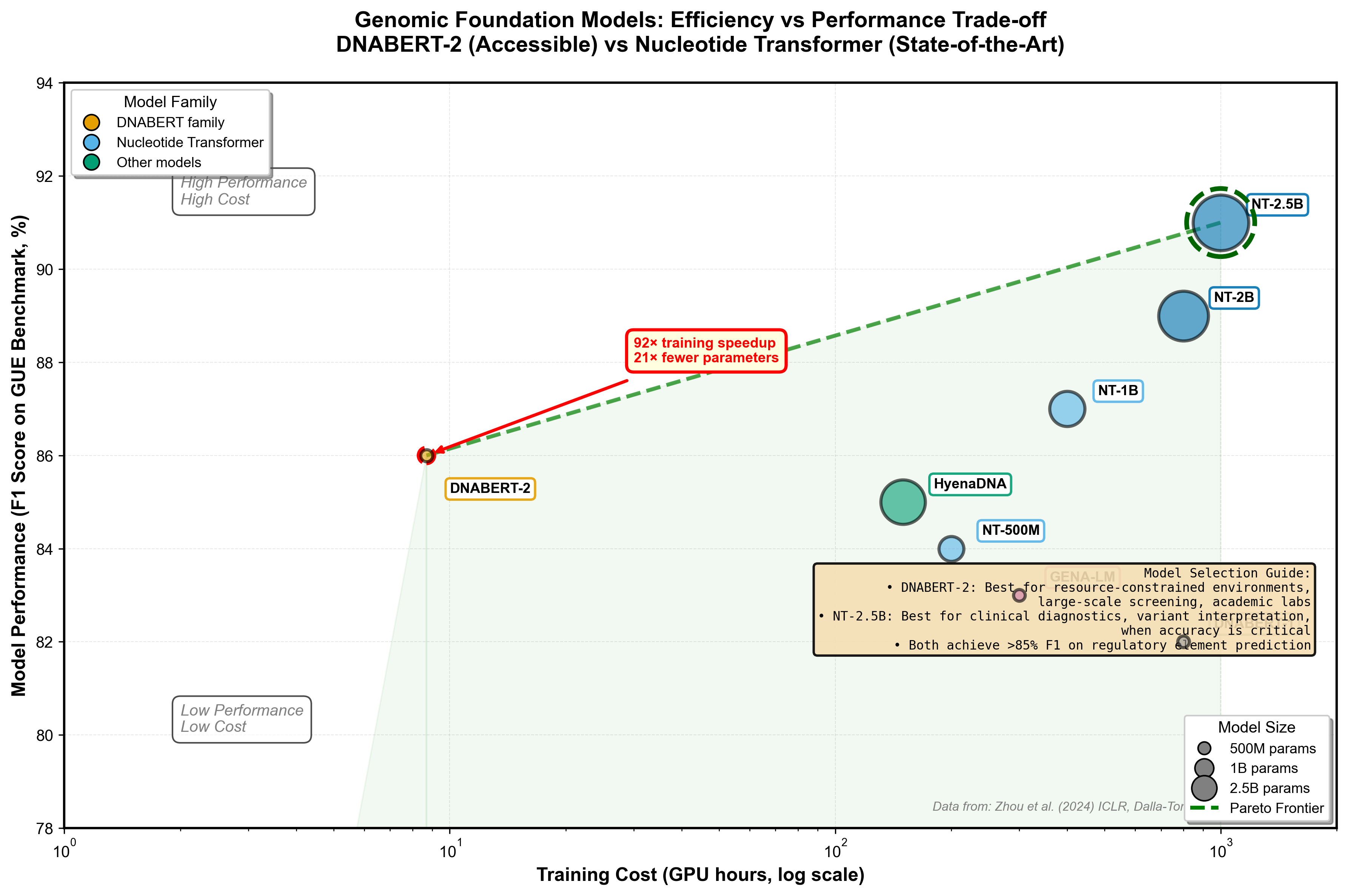
Figure 3: DNABERT-2 vs Nucleotide Transformer trade-off analysis. DNABERT-2's BPE tokenization achieves 92× faster training with competitive performance (efficiency corner), while Nucleotide Transformer 2.5B delivers peak accuracy at higher computational cost (performance corner). The Pareto frontier shows optimal choices depending on your constraints. Bubble sizes indicate model parameters (117M for DNABERT-2, 2.5B for NT). Data from GUE benchmark.
Model selection guide: Use DNABERT-2 for regulatory element analysis in human genomes when efficiency matters, especially for large-scale screening or when compute is constrained. Use Nucleotide Transformer 2.5B for highest-accuracy variant effect prediction, cross-species comparative genomics, or when every percentage point of accuracy matters (clinical diagnostics). For sequences longer than 10kb, consider HyenaDNA's exceptional runtime scalability.
[CODE_EXAMPLE: Using DNABERT-2 for regulatory element prediction]
# Install: pip install transformers torch
from transformers import AutoTokenizer, AutoModelForSequenceClassification
import torch
# Load DNABERT-2 fine-tuned on promoter classification
model_name = "zhihan1996/DNABERT-2-117M"
tokenizer = AutoTokenizer.from_pretrained(model_name, trust_remote_code=True)
model = AutoModelForSequenceClassification.from_pretrained(model_name, trust_remote_code=True)
# Example DNA sequence (200bp region)
dna_sequence = "ATCGATCGATCGATCGATCG" * 10 # Repeat for demo
# Tokenize with BPE (Byte Pair Encoding)
inputs = tokenizer(dna_sequence, return_tensors="pt", padding=True, truncation=True, max_length=512)
# Predict whether this sequence is a promoter
model.eval()
with torch.no_grad():
outputs = model(**inputs)
logits = outputs.logits
probabilities = torch.softmax(logits, dim=1)
is_promoter = probabilities[0][1].item() # Probability of being a promoter
print(f"Promoter probability: {is_promoter:.3f}")
# For regulatory element scanning, slide a window across genomic regions:
def scan_regulatory_elements(sequence, window_size=200, stride=50):
predictions = []
for i in range(0, len(sequence) - window_size + 1, stride):
window = sequence[i:i+window_size]
inputs = tokenizer(window, return_tensors="pt", truncation=True, max_length=512)
with torch.no_grad():
outputs = model(**inputs)
prob = torch.softmax(outputs.logits, dim=1)[0][1].item()
predictions.append((i, prob))
return predictions
# Returns list of (position, promoter_probability) tuples for the entire sequence
DNABERT-2's BPE tokenization learns biologically meaningful subsequences rather than fixed k-mers, capturing transcription factor binding motifs and regulatory patterns. The attention weights (accessible via outputs.attentions) reveal which genomic positions the model considers important for classification, providing interpretable insights into regulatory logic.
Small Molecules: SMILES as Molecular Language
Drug discovery requires predicting molecular properties before synthesis--toxicity, solubility, membrane permeability, metabolic stability. SMILES (Simplified Molecular Input Line Entry System) represents molecules as text strings, enabling NLP techniques for chemistry. But molecules present unique challenges: multiple valid SMILES for the same compound, 3D conformations affecting properties, and the need to predict dozens of properties simultaneously.
ChemBERTa: The Foundation
ChemBERTa adapted BERT architecture for SMILES strings, establishing the paradigm. Trained on ZINC (100k-250k molecules) and PubChem (77 million molecules--one of the largest molecular pre-training datasets), ChemBERTa learns chemical grammar: functional groups, bonding patterns, ring structures. Available on Hugging Face with multiple variants, it's the go-to baseline for molecular property prediction.
Citation: Chithrananda et al. (2020). ChemBERTa: Large-Scale Self-Supervised Pretraining for Molecular Property Prediction. arXiv.
MTL-BERT: Multitask Learning for Drug Development
MTL-BERT extends the foundation with three innovations. First, multitask learning: simultaneously train on 60 datasets (16 regression, 44 classification tasks) covering ADMET properties--Absorption, Distribution, Metabolism, Excretion, Toxicity. Second, SMILES enumeration: generate 20 different valid SMILES strings for each molecule as data augmentation, forcing the model to learn canonical representations. Third, interpretable attention: visualization reveals which molecular substructures the model focuses on for each property.
The interpretability matters. For water solubility predictions, MTL-BERT correctly prioritizes polar groups (hydroxyl, carboxyl). For mutagenicity, it identifies known structural alerts--nitro aromatics, epoxides. This chemical intuition builds trust when deploying models for real drug discovery decisions.
SMILES-based models excel at virtual screening--filtering millions of candidate molecules to find the few hundred worth synthesizing. For lead optimization--iteratively improving a drug candidate--they predict how structural modifications affect multiple properties simultaneously. The limitation: SMILES only capture 2D connectivity, not 3D conformations. For problems where shape matters (protein binding pockets), graph-based methods or direct 3D representations may work better.
When to use SMILES models: Early-stage drug discovery, virtual screening, multi-parameter optimization (want both potent and non-toxic), and ADMET predictions. Avoid for problems requiring explicit 3D structure (docking, conformational analysis) or for biologics (antibodies, protein therapeutics).
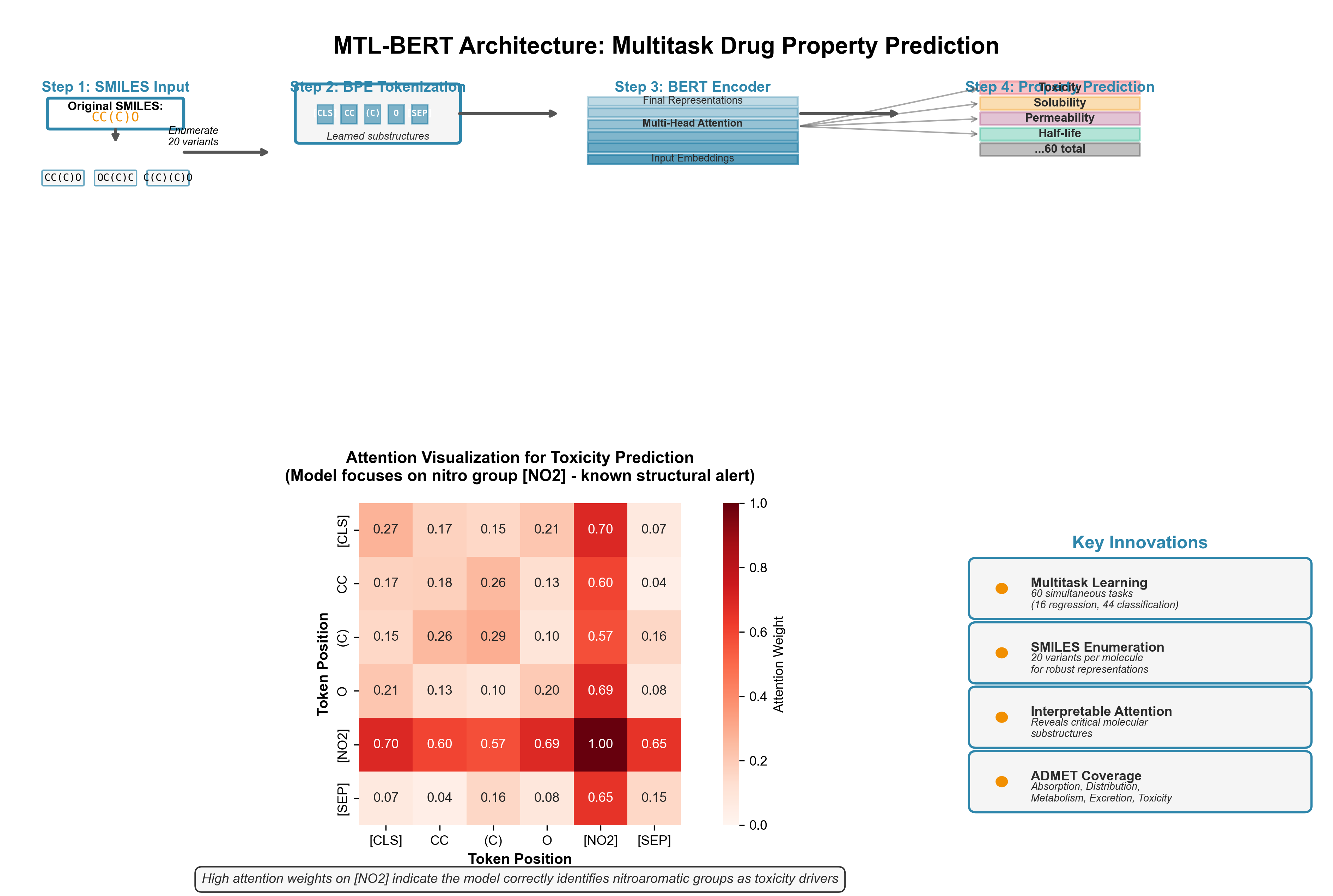
Figure 4: MTL-BERT architecture for multitask molecular property prediction. SMILES strings are tokenized using BPE, processed through a BERT encoder, and fed to 60 parallel prediction heads covering ADMET properties. The attention heatmap (bottom) shows interpretability--the model correctly focuses on nitro groups (red regions) when predicting mutagenicity/toxicity, matching known structural alerts in medicinal chemistry. SMILES enumeration (top right) demonstrates data augmentation where "CC(C)O" and "OC(C)C" represent the same isopropanol molecule.
Single-Cell & Multi-Omics: Understanding Cellular Complexity
Single-cell technologies measure gene expression, chromatin accessibility, protein levels, and spatial location in individual cells. Foundation models for single-cell data must handle millions of cells, thousands of genes, sparse measurements (most genes unexpressed in any given cell), and batch effects from experimental variation.
scGPT: GPT for Cells
scGPT applies the generative pre-trained transformer paradigm to single-cell biology. Pre-trained on 33 million single-cell RNA-seq profiles, it treats each cell as a "sentence" and genes as "words." Six major applications emerged from transfer learning: batch integration (correcting technical variation), multi-omic integration (linking RNA with protein measurements), cell-type annotation, perturbation prediction (what happens if you knock out a gene), gene network inference, and cell embedding for downstream analysis.
The "GPT for cells" framing isn't just marketing--the architecture and training objective directly parallel language models. Mask some genes, predict the missing expression values. The learned representations capture cellular states, developmental trajectories, and gene regulatory relationships.
Recent evaluations show nuance: scGPT achieves state-of-the-art performance on some tasks (batch correction, cell annotation with fine-tuning) but struggles with zero-shot generalization on others (clustering cells from completely new tissues). This honest assessment is important--foundation models aren't magic, and understanding their limitations prevents misapplication.
Citation: Cui et al. (2024). scGPT: toward building a foundation model for single-cell multi-omics using generative AI. Nature Methods, 21, 1470-1480.
scPRINT: Gene Networks from Attention
scPRINT scales to 50 million cells from CellxGene, making it the largest single-cell foundation model by training data. The architectural innovation is using attention matrices for gene regulatory network inference. Transformer attention weights reveal which genes the model considers when predicting others--a proxy for regulatory relationships.
scPRINT outperforms GENIE3 (a traditional network inference method) by 67% at recovering known gene-gene connections (verified in Nature Communications 2025). It shows enrichment for cell type-specific marker genes in attention patterns. And it achieves strong zero-shot performance on batch correction and cell annotation--tasks where scGPT required fine-tuning.
The integration with ESM2 protein embeddings reveals cross-domain foundation model synergy. By incorporating protein structure information (via ESM2) with gene expression patterns, scPRINT learns richer representations. A gene that codes for a protein kinase gets context from ESM2's knowledge of kinase structures, improving predictions about its regulatory role.
Citation: Heumos et al. (2024). scPRINT: pre-training on 50 million cells allows robust gene network predictions. bioRxiv.
scMRDR: The Multi-Omics Challenge
Measuring multiple molecular layers in the same cells--RNA, chromatin accessibility, surface proteins--provides comprehensive cellular views. But single-cell multi-omic data is almost always unpaired: you measure RNA in some cells, chromatin in others, not both in each cell. Integrating unpaired modalities is hard.
scMRDR uses a β-VAE (beta-Variational Autoencoder) architecture to disentangle modality-shared representations (cellular identity, state) from modality-specific representations (technical artifacts, measurement noise). Isometric regularization preserves biological heterogeneity during integration. The result: accurate modality alignment, batch correction, and biological signal preservation across unpaired datasets.
The framework scales to large datasets and handles more than two omics types--critical as technologies like CITE-seq (RNA + surface proteins), Multiome (RNA + chromatin), and spatial transcriptomics (RNA + location) become routine. scMRDR's NeurIPS 2024 Spotlight acceptance validates the approach's importance.
Citation: Sun et al. (2024). scMRDR: a scalable and flexible framework for unpaired single-cell multi-omics data integration. NeurIPS 2024 Spotlight.
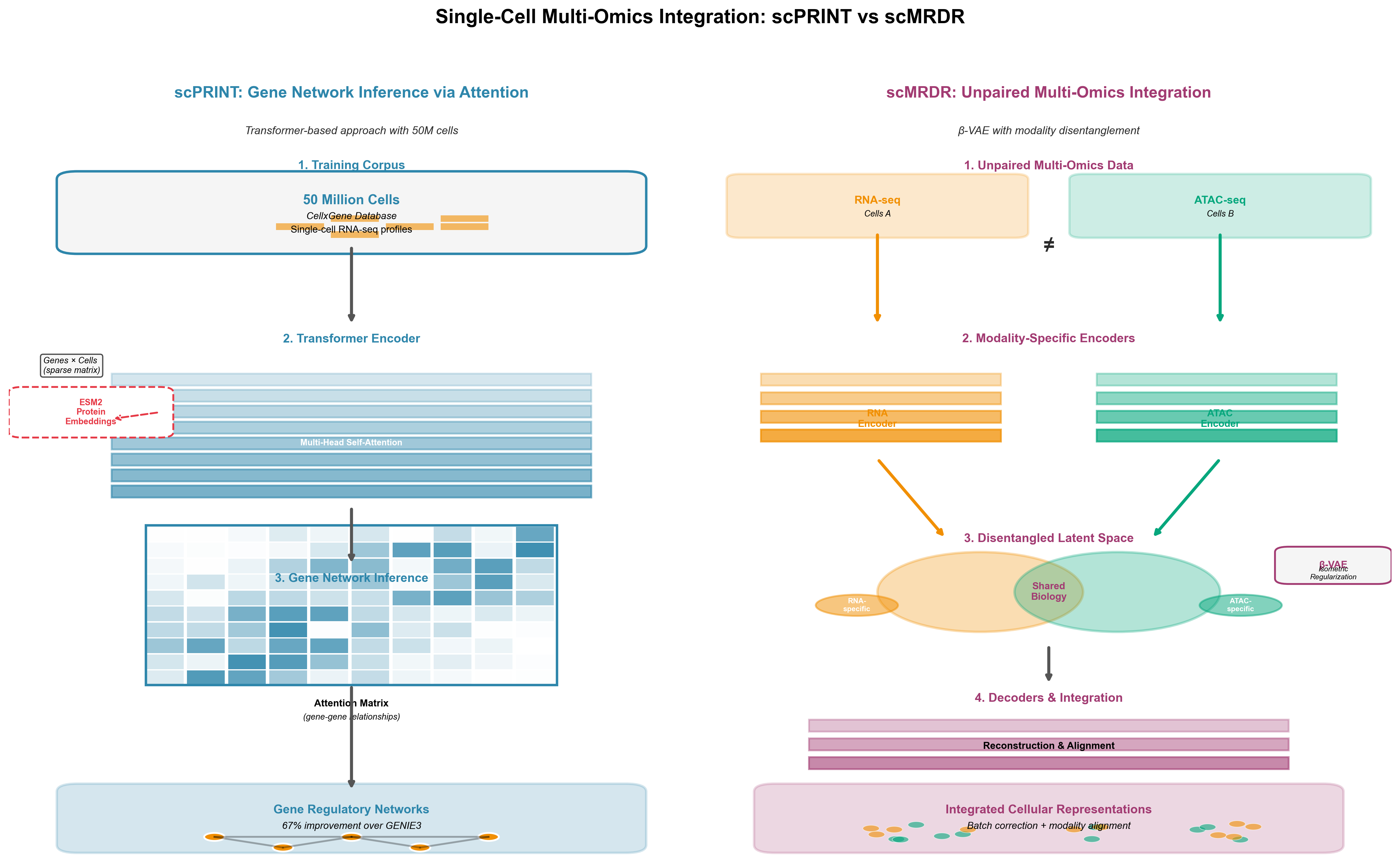
Figure 5: Two approaches to single-cell multi-omics integration. Left: scPRINT uses transformer attention on 50M cells to infer gene regulatory networks from attention patterns, achieving 67% better performance than traditional methods. Right: scMRDR employs β-VAE to integrate unpaired RNA and chromatin data by separating shared cellular identity from modality-specific technical artifacts. Both represent state-of-the-art for different integration challenges.
Framework selection: Use scGPT when you have large labeled datasets and want fine-tuned performance on specific tasks. Use scPRINT for gene network inference, zero-shot applications, or when you want to leverage protein structure information. Use scMRDR specifically for multi-omic integration problems where you need to align unpaired datasets.
Pathology: From Bench to Bedside
Digital pathology foundation models aim to automate cancer diagnosis, predict patient outcomes, and detect biomarkers from histopathology images. Unlike other biological domains where validation means comparing to databases, pathology models face the hardest test: do they work reliably in hospitals?
Atlas: State-of-the-Art in Two Months
Atlas, developed by Mayo Clinic, Charité, and Aignostics, trained on 1.2 million whole slide images from two major medical institutions. The rapid development timeline--less than two months from initiation to state-of-the-art--demonstrates what focused collaboration achieves. Atlas uses a ViT-H/14 (Vision Transformer) architecture with the RudolfV training paradigm, adapted for pathology-specific challenges.
The surprising result: Atlas achieves state-of-the-art performance across 21 public benchmarks despite being neither the largest model by parameters nor training data size. Some competing models trained on 44 terabytes of data or with more parameters perform worse. Why?
Data quality over quantity: Curated institutional data from two major medical centers, with diverse patient populations and staining protocols, beats indiscriminately collected web images. Multi-stain training: Atlas handles H&E (standard), immunohistochemistry (protein markers), and special stains (specific tissue components) in a unified model--real-world pathology labs use all of these. Clinical institution-led development: Pathologists guided development from day one, ensuring the model learns features relevant to diagnosis, not just benchmark performance.
Citation: Nechaev et al. (2025). A Novel Pathology Foundation Model by Mayo Clinic, Charité, and Aignostics. arXiv.
The Robustness Challenge
Recent research reveals foundation models' vulnerability to cross-institution variation. Staining procedures, scanner settings, tissue preparation methods, and patient populations vary between hospitals. A model performing well on Mayo Clinic slides might struggle with slides from a community hospital using different protocols.
Atlas addresses this through multi-institution training (Mayo + Charité) with different staining protocols and patient demographics. But the problem extends beyond training diversity. Clinical deployment requires continuous monitoring, recalibration, and validation on each institution's data. The gap between benchmark performance and reliable clinical use is the hardest problem in medical AI.
Clinical deployment barriers: Privacy regulations (HIPAA, GDPR) complicate data sharing. Hallucination risk--models generating plausible but incorrect diagnoses--is unacceptable in medicine. Regulatory pathways (FDA approval in the US) remain unclear for foundation models. Interpretability matters more than in other domains; pathologists need to understand why a model flagged a region as suspicious.
Atlas's development model--clinical institutions leading, not tech companies--provides a clearer path to regulatory approval. The Mayo Clinic digital pathology platform integrates Atlas into actual diagnostic workflows, enabling prospective validation studies that FDA requires.
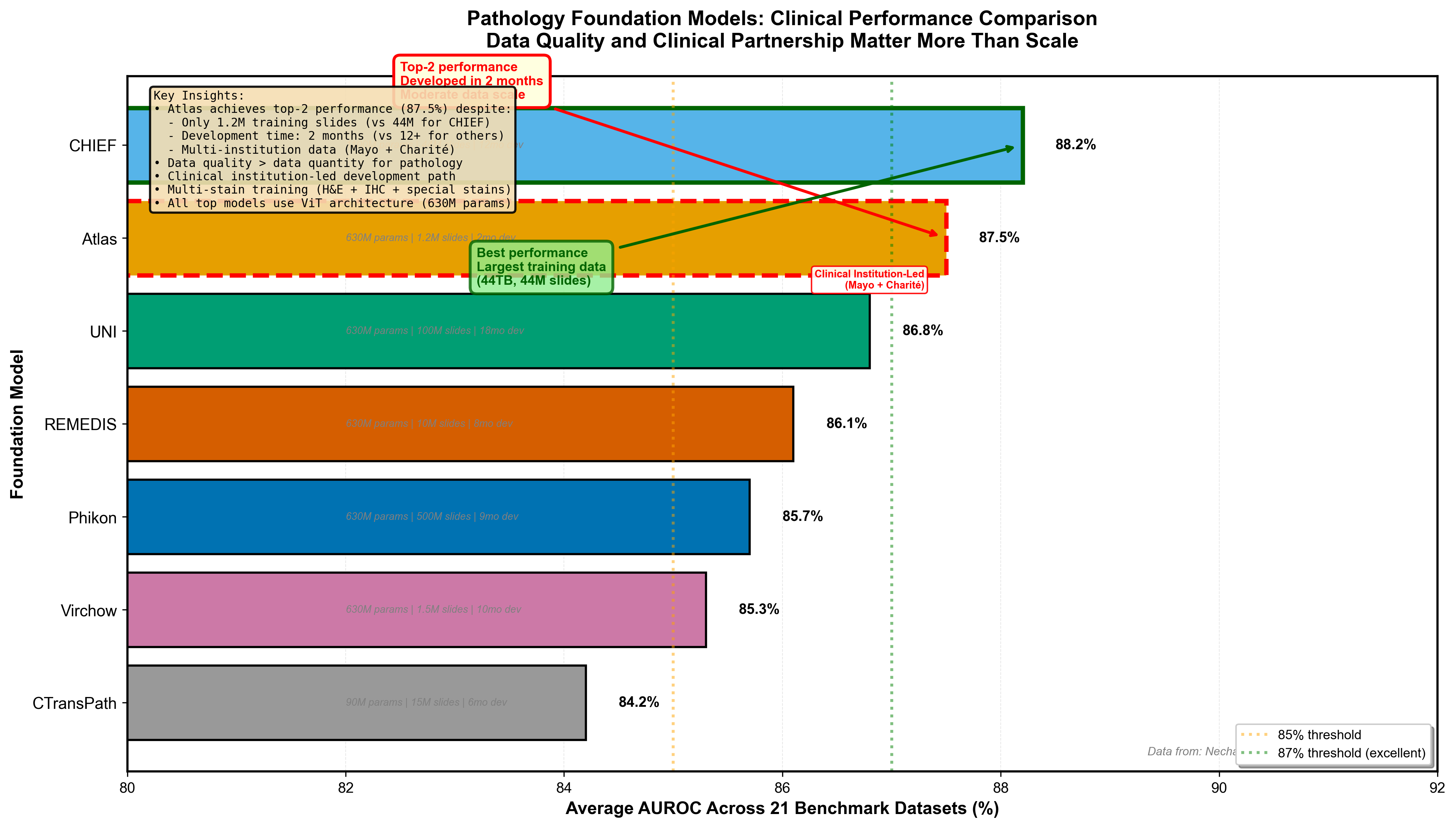
Figure 6: Pathology foundation model performance across 21 public benchmarks. Atlas achieves top-tier performance despite moderate scale (1.2M slides), demonstrating that data quality and clinical institution leadership matter more than raw scale. CHIEF trained on 44TB performs well but doesn't surpass Atlas. UNI and other models show varying performance. This pattern challenges the "bigger is always better" assumption--curation and clinical guidance during development prove decisive for medical AI. Data from Nechaev et al. (2025).
The Hard Problems: What Foundation Models Still Get Wrong
Beneath the impressive benchmark numbers, biological foundation models face three critical challenges that limit their reliability.
Interpretability: Understanding the Black Box
Biologists need to understand mechanisms, not just predictions. When scPRINT predicts a gene regulatory connection, does it reflect real biology or a statistical artifact? When MTL-BERT predicts a molecule is toxic, which substructure causes the problem?
Attention visualization helps--seeing which genomic regions or molecular features models focus on--but attention weights don't always correspond to causal relationships. Perturbation studies (systematically altering inputs to measure output changes) provide mechanistic insights but are computationally expensive. The fundamental tension: larger models with more parameters achieve better performance but are harder to interpret.
Biological applications demand interpretability more than most ML domains. A model that predicts a protein-protein interaction must explain what structural features drive the interaction, because biologists will design experiments to test those predictions. Without interpretability, foundation models remain hypothesis generators, not validated biological knowledge.
Hallucination: The Confidence Problem
Large language models hallucinate--generate plausible but false information. Biological foundation models do the same. AlphaFold3 predicts a protein structure with high confidence that turns out to be wrong when crystallized experimentally. scGPT predicts gene expression patterns in a cell type it never saw during training, but the predictions reflect training data biases, not real biology.
The danger is overconfidence. Models trained on millions of examples learn to predict with narrow error bars, even when extrapolating beyond their training distribution. Uncertainty quantification--estimating when predictions are unreliable--remains an open research problem.
For clinical applications, hallucination is disqualifying. A pathology model that confidently misdiagnoses cancer isn't just wrong--it's dangerous. Medical AI requires calibrated confidence: the model must know what it doesn't know.
Evaluation: Benchmarks Versus Biology
Standard ML metrics (accuracy, AUROC, F1 score) don't capture biological validity. A genomic model that perfectly predicts held-out test data might still fail on rare variants it never encountered. A drug discovery model that optimizes solubility might generate molecules impossible to synthesize.
The gap between benchmark performance and real-world utility is largest in clinical domains. Pathology models evaluated on curated benchmark datasets may not generalize to routine clinical cases with poor tissue quality, unusual staining, or rare cancer subtypes. DNABERT-2's GUE benchmark helps standardize genomics evaluation, but many biological questions lack established ground truth.
Experimental validation remains the ultimate test. Computational predictions must be verified in the lab--protein structures crystallized, predicted drug candidates synthesized and tested, inferred gene networks validated with genetic perturbations. Foundation models accelerate hypothesis generation, but they don't replace experiments.
What's Next: The Multimodal Future
Foundation models in biology are evolving along three frontiers.
Multimodal integration combines complementary data types. OmiCLIP integrates histopathology images with spatial transcriptomics, learning how tissue architecture relates to gene expression. Trained on 2.2 million tissue patches from 1,007 samples across 32 organs, it treats transcriptomics as a "sentence" of highly expressed genes paired with image patches. Med-PaLM Multimodal extends further, combining clinical text, medical images, and genomic data in a single generative vision-language model.
The promise: match how human experts reason--integrating patient history (text), radiology (images), pathology (images), and genomic sequencing (sequences) to reach diagnoses. The challenge: multimodal models are more complex, require diverse data, and are harder to interpret than unimodal models.
Spatial omics adds location information to molecular measurements. Nicheformer, pre-trained on 110 million cells (human and mouse, 73 organs/tissues) with both dissociated and image-based spatial data, learns tissue dependencies and spatial cellular representations. Understanding how cells organize in space--tumor microenvironments, immune cell infiltration, tissue architecture--is critical for understanding disease and designing therapies.
Generative capabilities extend beyond prediction to design. ProteinAE uses diffusion autoencoders to compress protein structures into latent spaces, enabling efficient generation of novel structures. Similar approaches for small molecules (molecular generation from latent spaces) and gene regulatory networks (designing synthetic circuits) promise to move from analyzing existing biology to creating new biology.
The field is moving fast. Models from 2024 will be superseded by 2025 developments. AlphaFold3's Nobel Prize arrival signals that foundation models for biology have reached maturity--deployed at scale, validated by the scientific establishment, and actively shaping drug discovery and biomedical research. But we're still in the early days. The hardest problems--clinical translation, interpretability, hallucination risk--remain unsolved.
Where to Go from Here
If you're an ML engineer considering biological applications, start with domains where your existing expertise transfers. NLP background? Genomic language models (DNABERT-2, Nucleotide Transformer) or SMILES-based drug discovery (ChemBERTa, MTL-BERT) are natural entry points. Computer vision experience? Pathology foundation models (Atlas) or spatial transcriptomics (OmiCLIP). The biological terminology is learnable; the ML fundamentals you already know apply.
For bioinformaticians evaluating which models to adopt: model selection depends on your constraints. Computational budget limited? DNABERT-2 and ESMFold prioritize efficiency. Need maximum accuracy? Nucleotide Transformer 2.5B and AlphaFold3 deliver peak performance at higher compute cost. Most importantly, use established benchmarks (GUE for genomics) and validate predictions experimentally--computational models accelerate discovery but don't replace bench work.
Computational biologists tracking the field: the shift from hypothesis-driven to data-driven biology is real, but it's not a replacement--it's a complement. Foundation models generate hypotheses at scale that would take years to develop manually. They reveal patterns in data too complex for traditional statistical methods. But they don't replace mechanistic understanding. The most powerful research combines foundation model insights with targeted experimental validation.
The foundation model revolution in biology isn't coming--it's here. AlphaFold3's Nobel Prize was the announcement. The next decade will determine whether these models fulfill their promise to accelerate drug discovery, personalize medicine, and deepen our understanding of life's molecular machinery. The early results are promising. The challenges are real. And the opportunity for technical practitioners to contribute is wide open.
We'll explore several models from this survey in depth in future posts--starting with AlphaFold3's diffusion architecture and its implications for drug discovery.
Resources
Open Source Models
- ESM-2/ESMFold: https://github.com/facebookresearch/esm
- DNABERT-2: https://github.com/MAGICS-LAB/DNABERT_2
- Nucleotide Transformer: https://github.com/instadeepai/nucleotide-transformer
- ChemBERTa: https://huggingface.co/seyonec/ChemBERTa-zinc-base-v1
- scGPT: Code available via Nature Methods publication
- AlphaFold3: https://github.com/google-deepmind/alphafold3 (non-commercial license)
Key Papers
- Abramson et al. (2024). AlphaFold 3. Nature, 630, 493-500. https://www.nature.com/articles/s41586-024-07487-w
- Lin et al. (2023). ESM-2. Science, 379, 1123-1130. https://www.science.org/doi/10.1126/science.ade2574
- Zhou et al. (2024). DNABERT-2. ICLR 2024. https://arxiv.org/abs/2306.15006
- Dalla-Torre et al. (2024). Nucleotide Transformer. bioRxiv. https://www.biorxiv.org/content/10.1101/2023.01.11.523679
- Cui et al. (2024). scGPT. Nature Methods, 21, 1470-1480. https://www.nature.com/articles/s41592-024-02201-0
- Sun et al. (2024). scMRDR. NeurIPS 2024 Spotlight. https://arxiv.org/abs/2510.24987
- Nechaev et al. (2025). Atlas. arXiv. https://arxiv.org/abs/2501.05409
- Chithrananda et al. (2020). ChemBERTa. arXiv. https://arxiv.org/abs/2010.09885
Benchmarks & Databases
- GUE: Genome Understanding Evaluation benchmark
- CellxGene: https://cellxgene.cziscience.com/
- ChEMBL: https://www.ebi.ac.uk/chembl/
- AlphaFold Database: https://alphafold.ebi.ac.uk/
- ESM Metagenomic Atlas: 617M predicted structures