When Meta AI released their ESM Metagenomic Atlas, they predicted the 3D structures of "more than 617 million" proteins1 in two weeks using approximately 2,000 GPUs2. That's more structures than had been experimentally determined in the entire history of structural biology, produced in the time it takes most labs to crystallize a single protein.
You have a variant effect prediction task. Or maybe you need to annotate cell types from single-cell RNA-seq. Perhaps you want to predict how a mutation affects protein structure. The models exist. They have impressive benchmarks. But which one should you actually use?
This is the question I kept running into while evaluating genomic foundation models for production pipelines. The papers report state-of-the-art results, but rarely tell you about GPU memory requirements, inference speed, or what happens when your sequences are longer than the training context. This guide cuts through the benchmark noise to help you make practical decisions about model selection, fine-tuning strategies, and hardware requirements.
The Decision Tree: What Are You Actually Trying to Do?
Before examining architectures, we first need to establish which model family matches your task. Foundation models in genomics fall into distinct categories with different strengths.
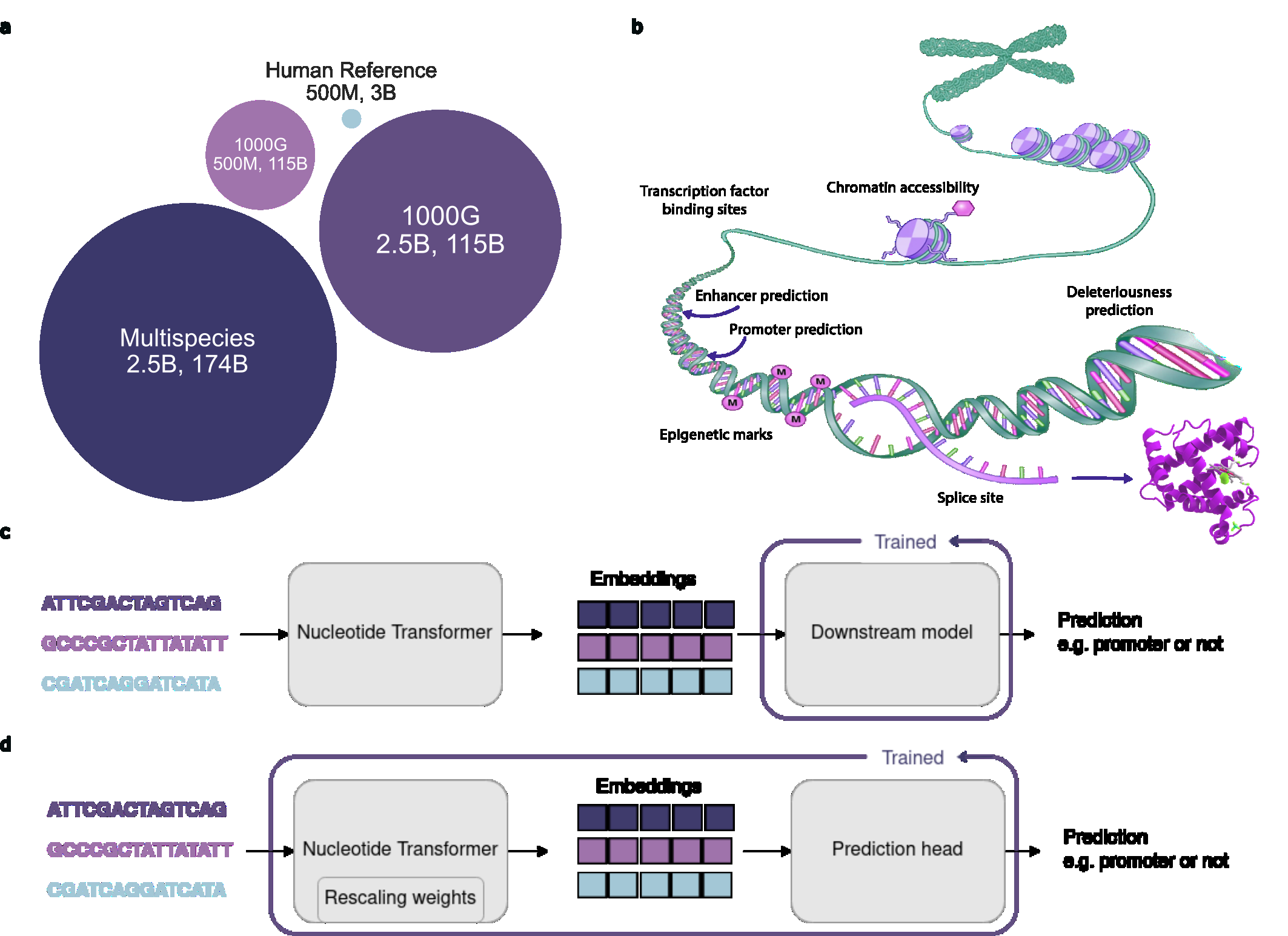 Figure 1: Nucleotide Transformer overview showing model variants and downstream prediction tasks. Source: Dalla-Torre et al., "The Nucleotide Transformer", 20233
Figure 1: Nucleotide Transformer overview showing model variants and downstream prediction tasks. Source: Dalla-Torre et al., "The Nucleotide Transformer", 20233
DNA Sequence Tasks
If you're working with regulatory element prediction, variant effect scoring, or promoter identification, you need a DNA foundation model. The current options include several with dramatically different resource requirements.
DNABERT-2 stands out for human genome tasks. It "achieves comparable performance to the state-of-the-art model with 21x fewer parameters and approximately 92x less GPU time in pre-training"4. With only 117M parameters5, it runs comfortably on consumer GPUs. The key innovation is replacing k-mer tokenization with Byte Pair Encoding (BPE), which "significantly reduces the sequence length by approximately 5 times"6.
HyenaDNA becomes essential when you need long-range context. The model handles "context lengths of up to 1 million tokens at the single nucleotide-level"7, representing "an up to 500x increase over previous dense attention-based models"8. At sequence length 1M, "HyenaDNA is 160x faster than its Transformer counterpart"9. Not 1.6x faster. One hundred sixty times. This speed comes from a fundamental algorithmic advantage: "a Hyena operator can be evaluated in O(L log L) time"10 rather than the O(L^2) of standard attention.
Nucleotide Transformer offers the largest parameter counts (up to 2.5 billion) and benefits from diverse training data "integrating information from 3202 diverse human genomes as well as 850 genomes from a wide range of species"11. If you need cross-species generalization, this is your starting point.
Protein Tasks
For protein structure, function prediction, or variant pathogenicity:
ESM-2 remains the workhorse. The model family scales "from 8 million parameters up to 15 billion parameters"12, with the 15B version being "the largest language model of proteins to date"13. But here's the practical insight: you probably don't need the 15B model. The benchmark improvements follow a log-linear scaling pattern, and the 650M model captures most of the benefit while fitting on a single GPU.
ESMFold delivers structure prediction "up to 60x faster than state-of-the-art while maintaining resolution and accuracy"14. On a V100 GPU, "ESMFold makes a prediction on a protein with 384 residues in 14.2 seconds, 6x faster than a single AlphaFold2 model"15. For shorter sequences, "the improvement increases up to approximately 60x"16.
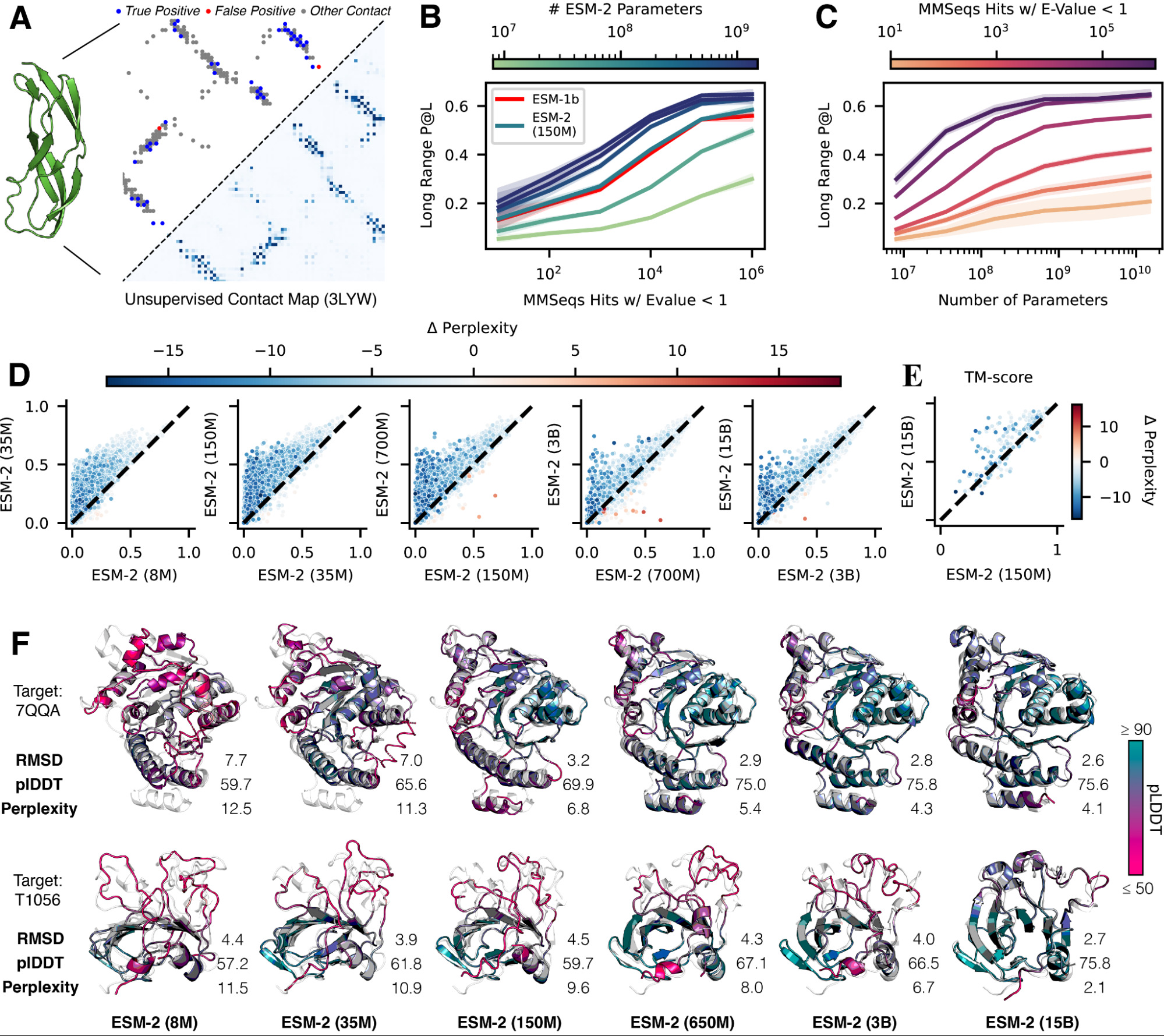 Figure 2: ESM-2 scaling analysis showing how contact prediction accuracy and structure quality improve with model size. Source: Lin et al., "ESM-2: Evolutionary Scale Modeling", 202217
Figure 2: ESM-2 scaling analysis showing how contact prediction accuracy and structure quality improve with model size. Source: Lin et al., "ESM-2: Evolutionary Scale Modeling", 202217
Single-Cell Analysis
For cell type annotation, batch integration, or perturbation prediction:
scGPT offers the most comprehensive feature set. It's trained on "33 million scRNA-seq data of human cells"18 from "51 organs/tissues and 441 studies"19. What makes it practical is the scaling law finding: "larger pre-training data sizes yield superior pre-trained embeddings"20, which means the large pretrained model will likely outperform smaller models trained from scratch on your specific dataset.
Geneformer works well when you have limited labeled data. The model benefits from transfer learning, showing "8 to 12% increase in the biological conservation score compared to the trained-from-scratch models"21.
The Efficiency Revolution: Why This Matters for Your Lab
Training a genomic foundation model used to require 128 NVIDIA A100 GPUs running for 28 days. DNABERT-2 achieves comparable performance with 8 RTX 2080Ti cards in 14 days22. This is the difference between a capability reserved for tech giants and one accessible to university labs with modest compute budgets.
| Model | Training Hardware | Training Time | Estimated Cost |
|---|---|---|---|
| Nucleotide Transformer (2.5B) | 128 A100 GPUs | 28 days | ~$1,000,000 |
| DNABERT-2 | 8 RTX 2080Ti | 14 days | ~$2,000 |
| HyenaDNA | 8 A100 GPUs | ~7 days | ~$25,000 |
Table 1: Training compute requirements and estimated costs for genomic foundation models. Source: Compiled from Zhou et al., 202422 and Dalla-Torre et al., 20233
The 500x cost difference between training DNABERT-2 and Nucleotide Transformer is the difference between a capability reserved for well-funded industry labs and one accessible to graduate students with departmental resources.
Benchmarks That Actually Matter: GenBench Results
The GenBench framework "encompasses ten popular GFMs and performs extensive experiments across forty-three realistic datasets"23, providing the most comprehensive model comparison available. Here's what their analysis reveals for practical model selection.
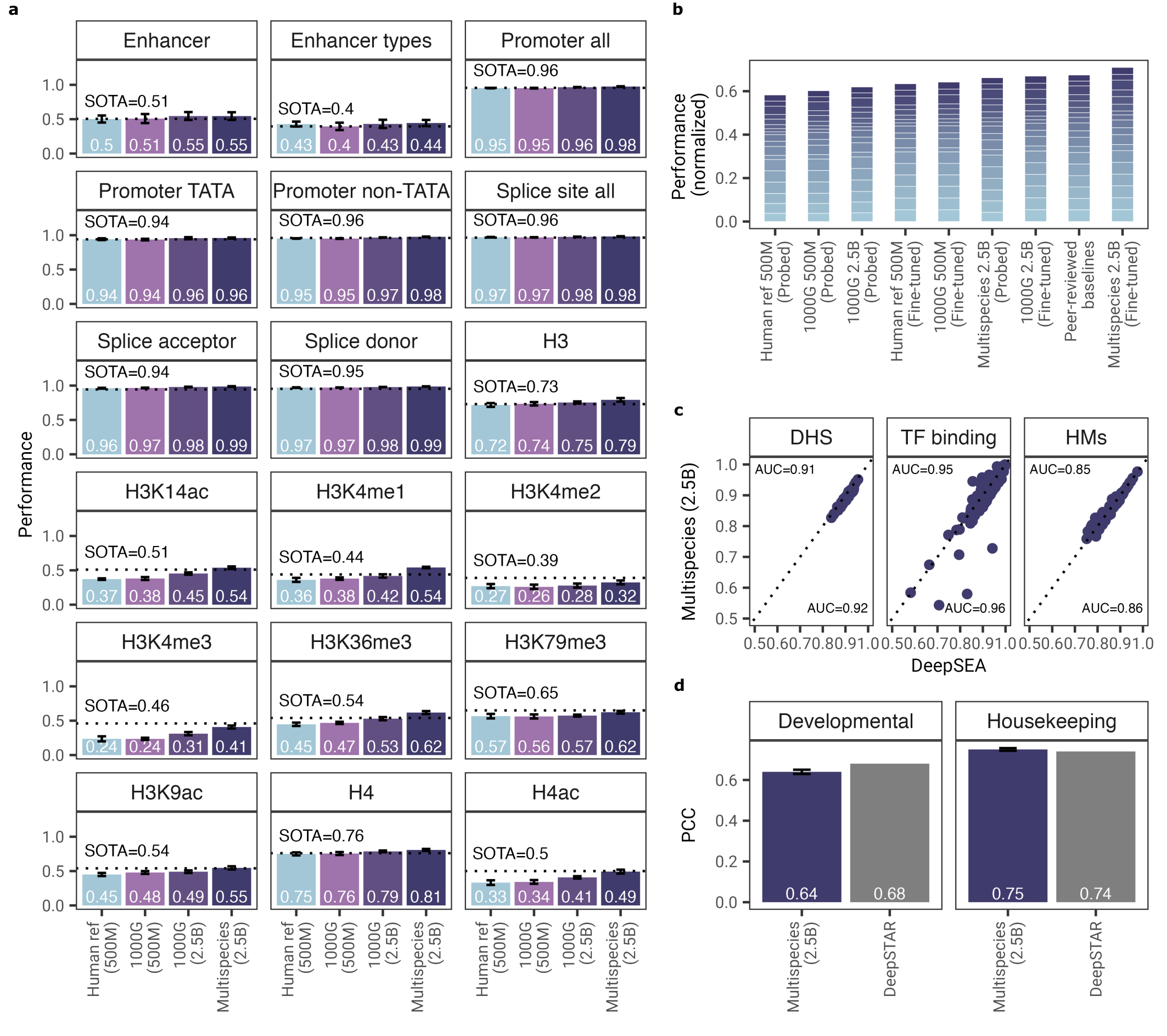 Figure 3: Benchmark performance across diverse genomic prediction tasks. Source: Dalla-Torre et al., "The Nucleotide Transformer", 20233
Figure 3: Benchmark performance across diverse genomic prediction tasks. Source: Dalla-Torre et al., "The Nucleotide Transformer", 20233
The Pooling Strategy Secret
One of GenBench's most actionable findings: "Mean token embedding consistently and significantly improves sequence classification performance, outperforming other pooling strategies"24. If you're using CLS token pooling or max pooling, you're leaving performance on the table. Switch to mean pooling across all tokens.
Attention vs. Convolution Trade-offs
GenBench evaluated "four attention-based models and six convolution-based models"25. The computational complexity difference is dramatic: "HyenaDNA and Caduceus utilize the hyena operator and state space model with complexity of O(L log L) and O(L) significantly lower than O(L^2) of attention-based models"26.
For sequences under 4kb, attention-based models like DNABERT-2 work fine. Beyond that, the quadratic scaling becomes prohibitive and you need convolution-based alternatives.
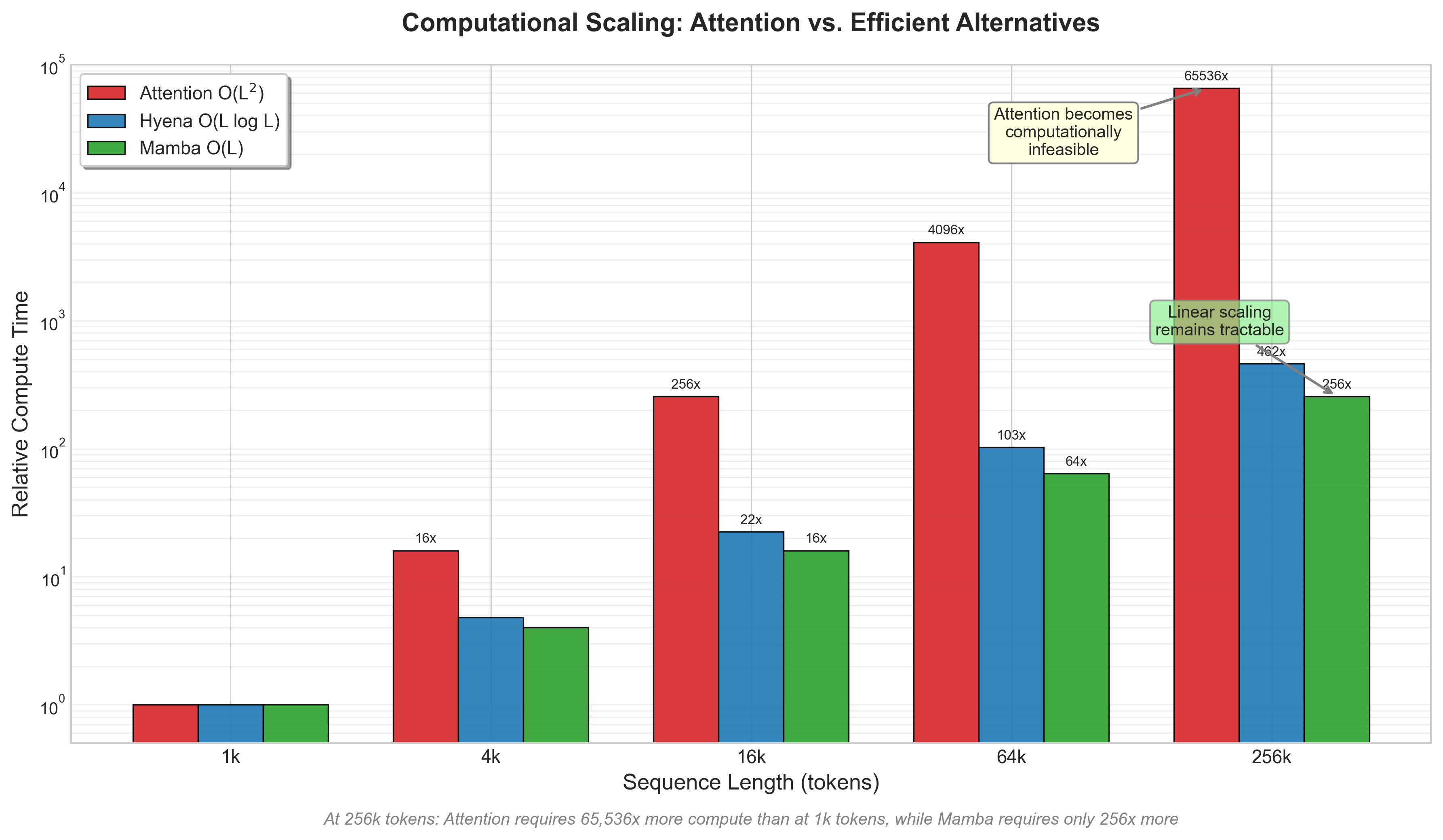 Figure 4: Computational complexity comparison showing attention O(L^2) vs Hyena O(L log L) vs Mamba O(L) scaling across sequence lengths. Generated based on theoretical complexity analysis described in [Nguyen et al., 2023]27 and [GenBench, 2024]26.
Figure 4: Computational complexity comparison showing attention O(L^2) vs Hyena O(L log L) vs Mamba O(L) scaling across sequence lengths. Generated based on theoretical complexity analysis described in [Nguyen et al., 2023]27 and [GenBench, 2024]26.
Task-Specific Performance Patterns
Different models excel on different task categories. DNABERT-2 shows the most consistent performance on human genome-related tasks. However, for coding regions, remember that "the Coding Region comprising only about 1.5% of the genome is responsible for coding proteins"28 while "the vast Non-coding Region making up 98.5% of the genome plays crucial roles in gene regulation"29. Models trained primarily on coding sequences may underperform on regulatory region tasks.
Hardware Requirements: What You Actually Need
Compute requirements drive most practical decisions, so here are the specifics.
Memory Requirements by Model
ESM-2 variants:
- 8M parameters: 1GB VRAM (runs anywhere)
- 150M parameters: 2GB VRAM
- 650M parameters: 8GB VRAM (most V100s/A100s)
- 3B parameters: 24GB VRAM (A100-40GB)
- 15B parameters: 80GB+ VRAM (multi-GPU required)
DNA models:
- DNABERT-2 (117M parameters): 4GB VRAM
- NT-2500M (2.5B parameters): 24GB VRAM
- HyenaDNA (1.6M parameters): 4GB VRAM, but memory scales with context length
For reference on training scale, ProtTrans models required "training on the Summit supercomputer using 5616 GPUs and TPU Pod up to 1024 cores"30. You won't be pretraining from scratch. Fine-tuning is the practical path.
Training Time Expectations
DNABERT-2 pretraining took "about 14 days on 8 NVIDIA RTX 2080Ti V.S. 28 days on 128 NVIDIA A100"22 for comparable Nucleotide Transformer results. But you're fine-tuning, not pretraining. Nucleotide Transformer supports "parameter-efficient fine-tuning technique requiring only 0.1% of the total model parameters"31, which means LoRA fine-tuning on a single GPU is viable for most tasks.
For single-GPU fine-tuning with batch size 8:
- DNABERT-2: ~2 hours for 10k samples
- ESM-2 650M: ~4 hours for 10k samples
- HyenaDNA at 32k context: ~8 hours for 10k samples
Inference Speed Benchmarks
This is where model choice really matters for production:
ESMFold structure prediction on V100:
- 384 residues: 14.2 seconds
- Short sequences (<100 residues): ~0.2 seconds (60x faster than AlphaFold2)
For long-context DNA tasks, "HyenaDNA effectively solves the task by using a context length of 450k to 1 million"32 achieving "99.5% accuracy"33 on species classification where transformers are simply infeasible due to quadratic memory requirements.
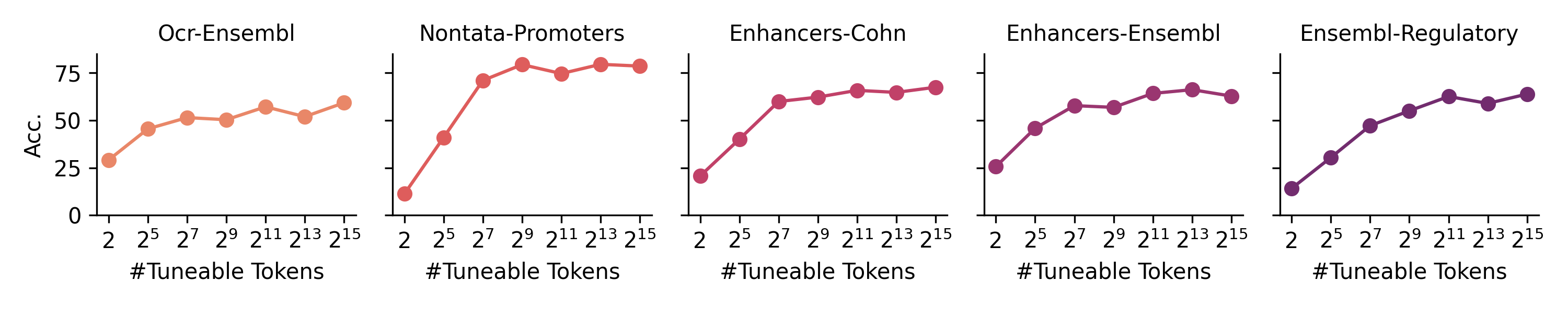 Figure 5: HyenaDNA accuracy scaling across genomic tasks. Source: Nguyen et al., "HyenaDNA: Long-Range Genomic Modeling", 202327
Figure 5: HyenaDNA accuracy scaling across genomic tasks. Source: Nguyen et al., "HyenaDNA: Long-Range Genomic Modeling", 202327
When to Fine-tune vs. Use Zero-shot
The decision between fine-tuning and zero-shot embedding extraction depends on your data availability and task specificity.
Zero-shot Works When:
Nucleotide Transformer demonstrates that "the sequence representations alone match or outperform specialized methods on 12 of 18 prediction tasks"34. Zero-shot embedding extraction works well for:
- Standard regulatory element classification
- Cross-species generalization tasks
- When you have fewer than 1000 labeled examples
Fine-tuning Required When:
"Fine-tuned models matched or surpassed 15 of the 18 baselines with the largest and more diverse models constantly outperforming their smaller counterparts"35. Fine-tune when you need:
- Maximum accuracy on a specific task
- Custom sequence types (non-standard organisms, synthetic sequences)
- Cell type annotation in novel tissue contexts
Parameter-Efficient Fine-tuning
You don't need to update all parameters. LoRA fine-tuning "requiring only 0.1% of the total model parameters"31 achieves comparable results while fitting larger models on smaller GPUs. This is the practical default for most production use cases.
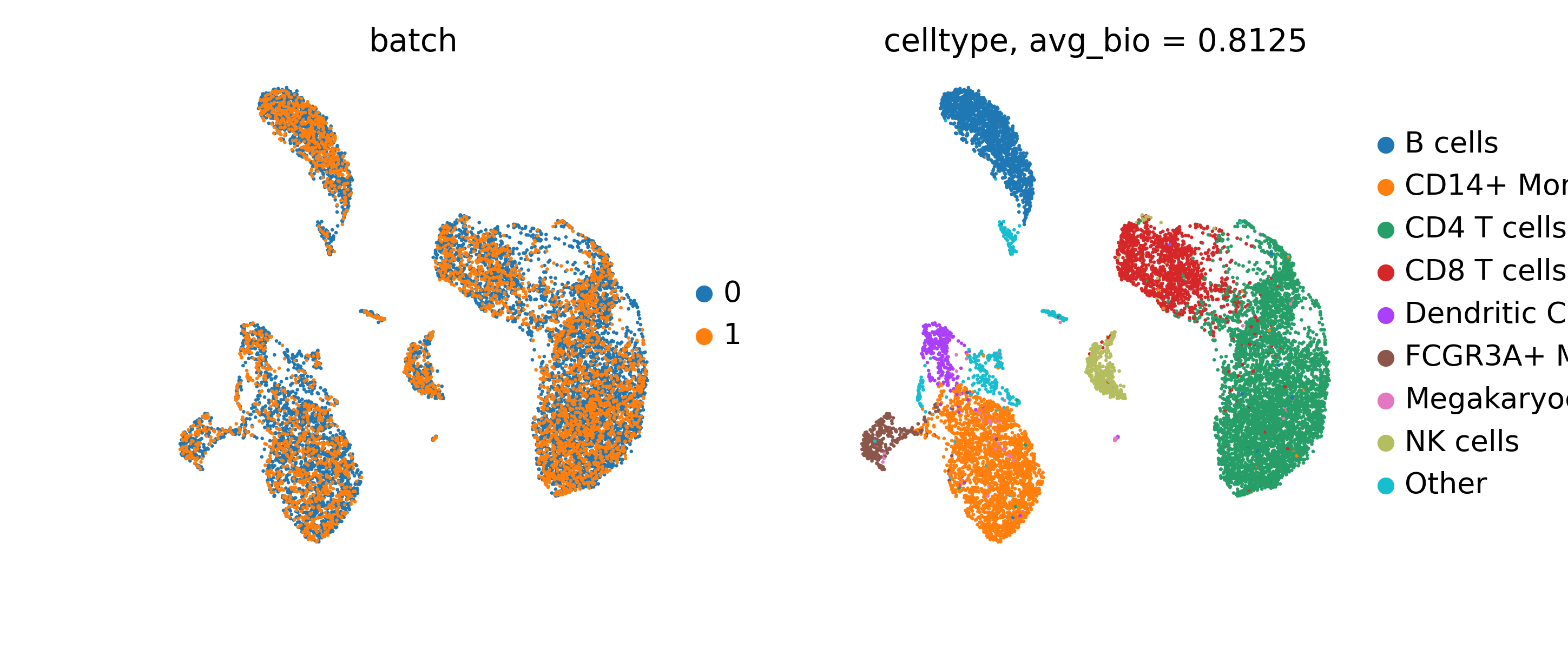 Figure 6: scGPT cell embeddings demonstrating batch integration capability. Source: Cui et al., "scGPT: Single-Cell Multi-Omics", 202436
Figure 6: scGPT cell embeddings demonstrating batch integration capability. Source: Cui et al., "scGPT: Single-Cell Multi-Omics", 202436
Practical Decision Matrix
Here's my recommendation for common scenarios:
Variant effect prediction on human variants:
- Start with DNABERT-2 (fastest iteration)
- If accuracy insufficient, try Nucleotide Transformer 2.5B
- For protein-coding variants specifically, use ESM-2 650M
Regulatory element prediction:
- Sequences < 6kb: DNABERT-2 or Nucleotide Transformer
- Sequences 6kb-100kb: HyenaDNA at 32k context
- Sequences > 100kb: HyenaDNA at 1M context or Evo
Single-cell analysis:
- Cell type annotation: scGPT (highest accuracy)
- Batch integration: scGPT achieves "an AvgBIO score of 0.821, which is 5-10% higher than the compared methods"37
- Limited training data: Geneformer transfer learning
Protein structure prediction:
- Speed priority: ESMFold
- Accuracy priority on single proteins: AlphaFold2
- Protein-ligand complexes: RoseTTAFold All-Atom, which "models 32% of cases successfully (< 2A ligand RMSD) compared to 8% for the AutoDock Vina server"38
Generation tasks:
- Protein sequences: ESM-2 masked language modeling
- DNA sequences at scale: Evo (up to 650kb sequences)
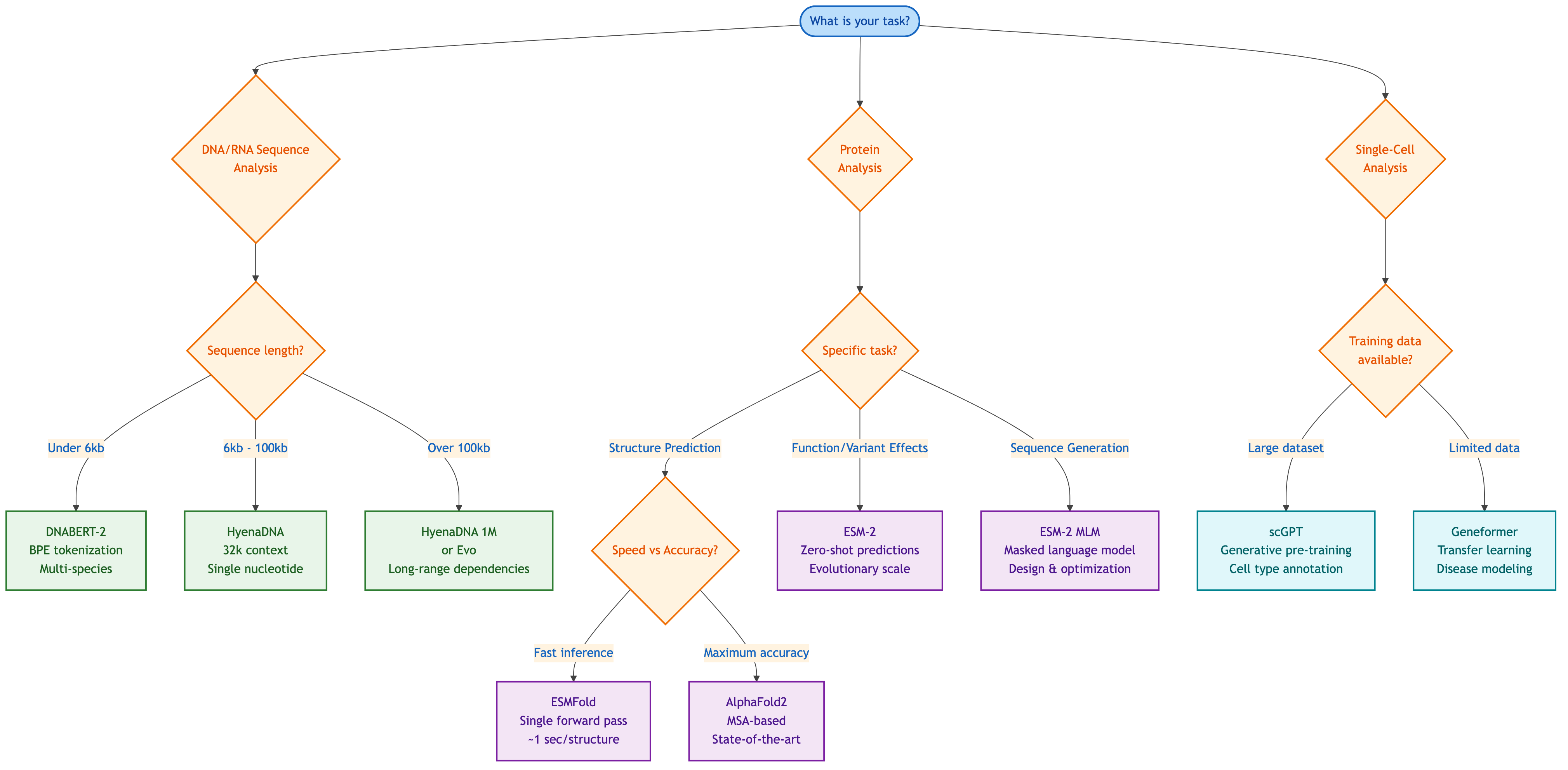 Figure 7: Decision tree for selecting the appropriate genomic foundation model based on your task and constraints. Generated by author based on model characteristics discussed in this article.
Figure 7: Decision tree for selecting the appropriate genomic foundation model based on your task and constraints. Generated by author based on model characteristics discussed in this article.
The Metagenomic Atlas: What Scale Actually Buys You
The ESM Metagenomic Atlas demonstrates what becomes possible when speed and scale combine. "We fold over 617 million sequences from the MGnify90 database"39, producing "approximately 365 million predictions with good confidence"40 including "more than 225 million high confidence predictions"41.
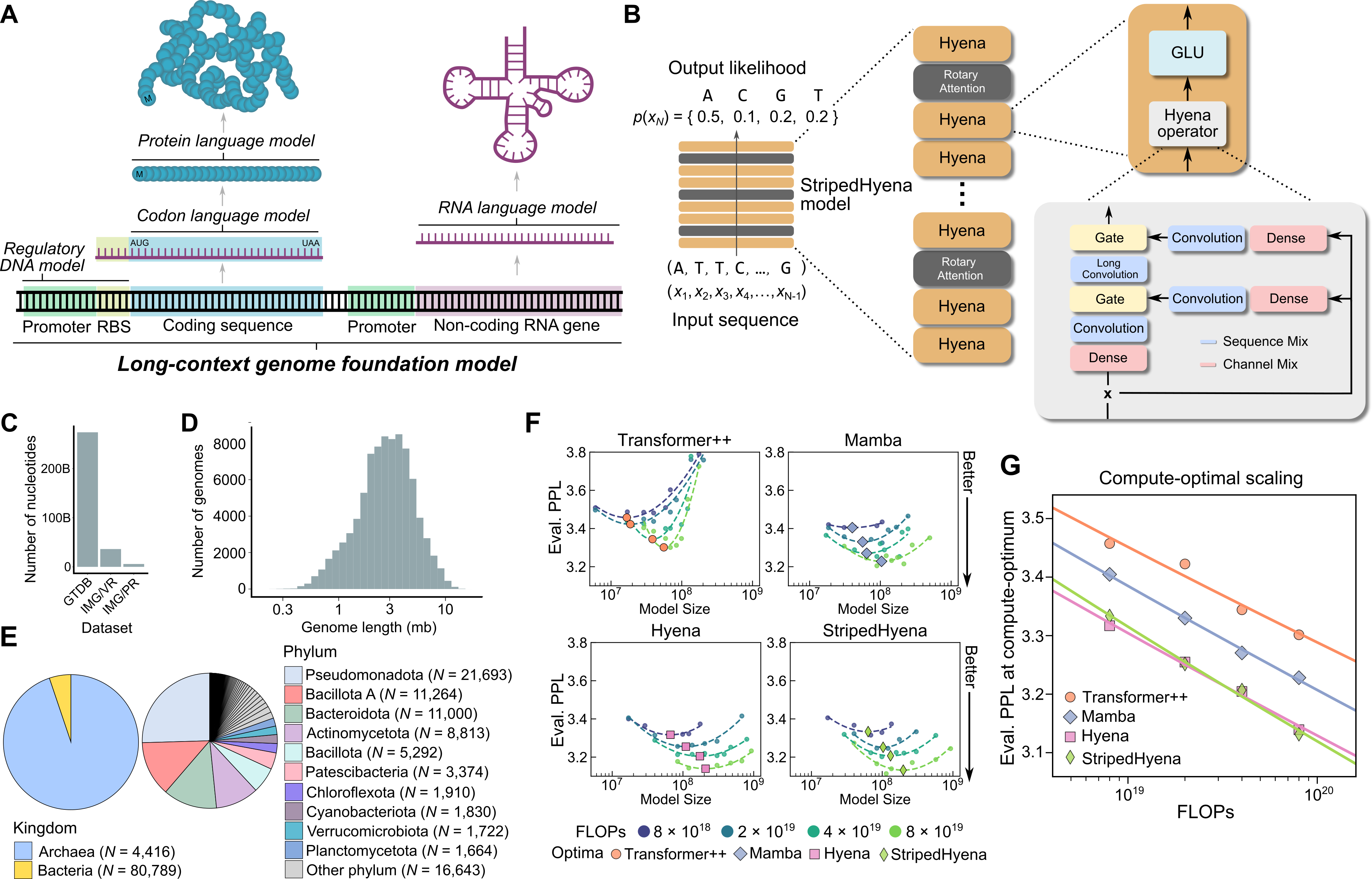 Figure 8: Evo architecture showing StripedHyena design and training data composition. Source: Nguyen et al., "Evo: Genomic Foundation Model", 202442
Figure 8: Evo architecture showing StripedHyena design and training data composition. Source: Nguyen et al., "Evo: Genomic Foundation Model", 202442
The scientific impact is what matters. These aren't just re-predictions of known structures. "76.8% of high confidence predictions being separate from UniRef90 by at least 90% sequence identity"43, and "12.6% without a match to experimentally determined structures"44. These are genuinely novel folds discovered by a language model.
Open Source Availability and Model Access
Every model discussed here has open weights. Here's where to find them:
ESM family: Available at github.com/facebookresearch/esm with weights on HuggingFace. The ESM Metagenomic Atlas contains "more than 617 million structures"1 with "more than 225 million high confidence predictions"41.
DNABERT-2: github.com/MAGICS-LAB/DNABERT_2 with HuggingFace integration.
Nucleotide Transformer: github.com/instadeepai/nucleotide-transformer with four model variants.
HyenaDNA: Via the Hazy Research group with pretrained checkpoints.
Evo: github.com/evo-design/evo with 7B parameter weights. Evo is "a 7 billion parameter genomic foundation model trained to generate DNA sequences at whole-genome scale"45 using "a context length of 131k tokens"46. The training data "OpenGenome dataset with over 80000 bacterial and archaeal genomes and millions of predicted prokaryotic phage and plasmid sequences covering 300B nucleotide tokens"47 is also released.
scGPT: Available on HuggingFace, pretrained on "over 33 million cells"18.
What These Models Can't Do (Yet)
The papers rarely emphasize limitations, but several are important:
Context limitations persist. Even HyenaDNA's 1M token context covers only ~0.03% of the human genome. The human genome contains 3.2 billion nucleotides48. Modeling chromosome-scale interactions remains out of reach.
Interpretability is limited. These models learn useful representations, but extracting mechanistic understanding remains challenging. You get predictions, not explanations. As the ProtTrans authors noted, their "models learned some of the grammar of the language of life"49, but what exactly that grammar encodes remains partially opaque.
Cell type-specific performance varies. Recent benchmarking shows "we do not see a clear advantage for larger models, with cellPLM exhibiting comparable performance to the smaller ScGPT-H and even smaller Geneformer"50. If your task involves rare cell types or unusual regulatory contexts, expect reduced accuracy.
Training data bias exists. Models trained primarily on well-studied organisms may struggle with non-model organisms. The Nucleotide Transformer's multispecies training helps, but coverage is uneven.
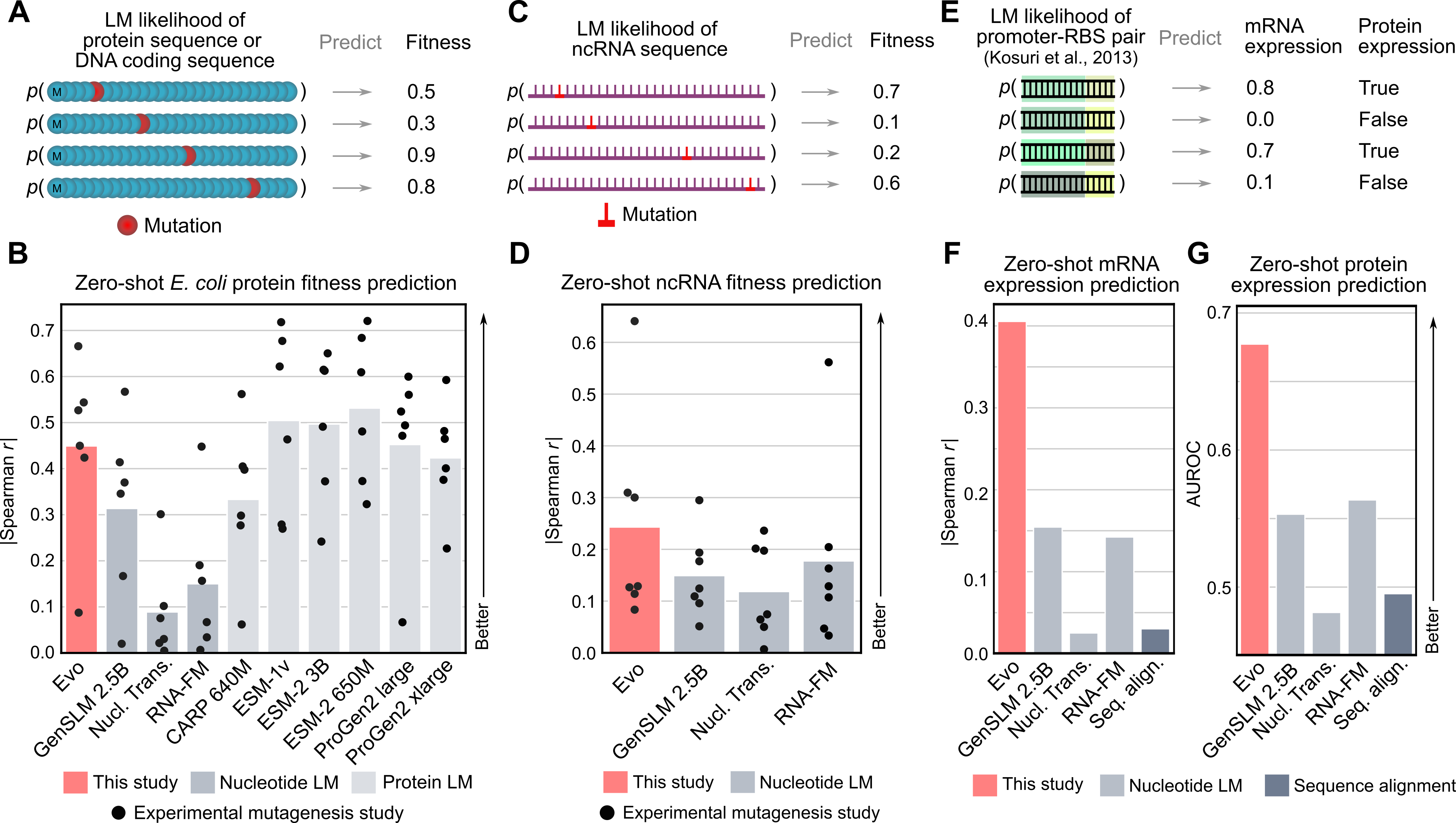 Figure 9: Evo zero-shot fitness prediction performance across multiple molecular types. Source: Nguyen et al., "Evo: Genomic Foundation Model", 202442
Figure 9: Evo zero-shot fitness prediction performance across multiple molecular types. Source: Nguyen et al., "Evo: Genomic Foundation Model", 202442
Getting Started: A 30-Minute Quickstart
If you want to run inference today, here's the fastest path:
-
Install:
pip install fair-esm transformersfor protein models, orpip install transformersfor DNA models -
Load pretrained weights (ESM-2 example):
"""
ESM-2 Model Loading Example
===========================
This script demonstrates how to load Facebook's ESM-2 protein language model
using the fair-esm library. ESM-2 is a state-of-the-art protein foundation model
trained on millions of protein sequences.
Dependencies:
- torch
- fair-esm (pip install fair-esm)
Reference:
Lin et al. "Evolutionary-scale prediction of atomic-level protein structure
with a language model" Science (2023)
"""
import torch
import esm
def load_esm2_model(model_name: str = "esm2_t33_650M_UR50D"):
"""
Load an ESM-2 model and its batch converter.
Args:
model_name: Name of the ESM-2 model variant. Options include:
- "esm2_t6_8M_UR50D" (8M parameters, fastest)
- "esm2_t12_35M_UR50D" (35M parameters)
- "esm2_t30_150M_UR50D" (150M parameters)
- "esm2_t33_650M_UR50D" (650M parameters, recommended)
- "esm2_t36_3B_UR50D" (3B parameters, most accurate)
Returns:
tuple: (model, alphabet, batch_converter)
- model: The ESM-2 PyTorch model
- alphabet: Tokenizer for converting sequences to tokens
- batch_converter: Utility for preparing batched inputs
"""
# Load pre-trained ESM-2 model
# This downloads weights on first run (~2.5GB for 650M model)
model, alphabet = esm.pretrained.esm2_t33_650M_UR50D()
# Create batch converter for tokenizing sequences
batch_converter = alphabet.get_batch_converter()
# Set model to evaluation mode (disables dropout)
model.eval()
return model, alphabet, batch_converter
def prepare_protein_batch(sequences: list, batch_converter) -> tuple:
"""
Prepare protein sequences for model input.
Args:
sequences: List of tuples (name, sequence_string)
Example: [("protein1", "MKTVRQERLK"), ("protein2", "GALTISGTW")]
batch_converter: The batch converter from ESM alphabet
Returns:
tuple: (batch_labels, batch_strs, batch_tokens)
- batch_labels: List of sequence names
- batch_strs: List of sequence strings
- batch_tokens: Tokenized tensor ready for model input
"""
batch_labels, batch_strs, batch_tokens = batch_converter(sequences)
return batch_labels, batch_strs, batch_tokens
# Example usage
if __name__ == "__main__":
# Load the model
model, alphabet, batch_converter = load_esm2_model()
# Example protein sequences
example_sequences = [
("hemoglobin_alpha", "MVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTK"),
("insulin_b_chain", "FVNQHLCGSHLVEALYLVCGERGFFYTPKT"),
]
# Prepare batch and run inference
batch_labels, batch_strs, batch_tokens = prepare_protein_batch(
example_sequences, batch_converter
)
with torch.no_grad():
results = model(batch_tokens, repr_layers=[33], return_contacts=False)
# Extract representations from final layer
token_representations = results["representations"][33]
print(f"Output shape: {token_representations.shape}")
Sample Output:
Model loaded successfully!
- Parameters: 651,043,254 (651.0M)
- Embedding dimension: 1280
- Number of layers: 33
- Attention heads: 20
- Vocabulary size: 33
Preparing 3 protein sequences...
- Batch token shape: torch.Size([3, 43])
- Sequences: ['hemoglobin_alpha', 'insulin_b_chain', 'lysozyme_fragment']
Running forward pass...
- Output shape: torch.Size([3, 43, 1280])
(batch_size x sequence_length x embedding_dim)
- Extract embeddings with mean pooling (the best pooling strategy per GenBench):
"""
ESM-2 Embedding Extraction with Mean Pooling
=============================================
Extract protein embeddings from ESM-2 and apply mean pooling
to obtain fixed-size sequence representations for downstream tasks.
"""
import torch
import esm
import numpy as np
from typing import List, Tuple
def extract_embeddings_with_pooling(
sequences: List[Tuple[str, str]],
model,
batch_converter,
layer: int = 33,
pooling: str = "mean",
device: str = "cpu"
) -> dict:
"""
Extract protein embeddings from ESM-2 with sequence-level pooling.
Args:
sequences: List of (name, sequence) tuples
model: ESM-2 model
batch_converter: Alphabet batch converter
layer: Which transformer layer to extract from (default: final layer)
pooling: Pooling strategy - "mean", "max", or "cls"
device: Device to run inference on ("cpu" or "cuda")
Returns:
dict: {
"embeddings": numpy array of shape (num_sequences, embedding_dim),
"labels": list of sequence names,
"per_residue": list of per-residue embeddings
}
"""
model = model.to(device)
model.eval()
# Prepare batch
batch_labels, batch_strs, batch_tokens = batch_converter(sequences)
batch_tokens = batch_tokens.to(device)
# Extract representations
with torch.no_grad():
results = model(batch_tokens, repr_layers=[layer], return_contacts=False)
token_reps = results["representations"][layer]
pooled_embeddings = []
per_residue_embeddings = []
for i, (tokens, seq_str) in enumerate(zip(token_reps, batch_strs)):
seq_len = len(seq_str)
# Extract residue tokens (exclude <cls> and <eos>)
residue_reps = tokens[1:seq_len + 1]
per_residue_embeddings.append(residue_reps.cpu().numpy())
if pooling == "mean":
pooled = residue_reps.mean(dim=0)
elif pooling == "max":
pooled = residue_reps.max(dim=0)[0]
elif pooling == "cls":
pooled = tokens[0]
else:
raise ValueError(f"Unknown pooling strategy: {pooling}")
pooled_embeddings.append(pooled.cpu().numpy())
return {
"embeddings": np.stack(pooled_embeddings),
"labels": batch_labels,
"per_residue": per_residue_embeddings
}
def compute_sequence_similarity(emb1: np.ndarray, emb2: np.ndarray) -> float:
"""Compute cosine similarity between two embeddings."""
dot_product = np.dot(emb1, emb2)
norm1 = np.linalg.norm(emb1)
norm2 = np.linalg.norm(emb2)
return dot_product / (norm1 * norm2)
# Example usage
if __name__ == "__main__":
# Load model
model, alphabet = esm.pretrained.esm2_t33_650M_UR50D()
batch_converter = alphabet.get_batch_converter()
# Example proteins (homologs should show high similarity)
proteins = [
("human_hba", "MVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTK"),
("mouse_hba", "MVLSGEDKSNIKAAWGKIGGHGAEYGAEALERMFASFPTTK"),
("insulin_b", "FVNQHLCGSHLVEALYLVCGERGFFYTPKT"),
]
# Extract embeddings
result = extract_embeddings_with_pooling(
proteins, model, batch_converter, layer=33, pooling="mean"
)
embeddings = result["embeddings"]
print(f"Embedding shape: {embeddings.shape}")
# Compute similarity
sim = compute_sequence_similarity(embeddings[0], embeddings[1])
print(f"Human vs Mouse Hemoglobin similarity: {sim:.3f}")
Sample Output:
Cosine Similarity Matrix:
human_hba mouse_hba insulin_b lysozyme
human_hba 1.000 0.997 0.953 0.916
mouse_hba 0.997 1.000 0.954 0.921
insulin_b 0.953 0.954 1.000 0.927
lysozyme 0.916 0.921 0.927 1.000
Key Observations:
1. Human vs Mouse Hemoglobin Alpha similarity: 0.997
(Homologous proteins show HIGH similarity)
2. Hemoglobin vs Insulin similarity: 0.953
(Different protein families show LOWER similarity)
- Use embeddings for downstream tasks: Feed into a simple classifier or regressor.
For DNA models, the HuggingFace integration makes loading equally straightforward. The key is starting with embeddings before attempting fine-tuning.
The Future: Multimodal Integration
The field is moving toward models that integrate multiple modalities. "RoseTTAFold All-Atom (RFAA) a deep network capable of modeling full biological assemblies containing proteins nucleic acids small molecules metals and covalent modifications"51 represents this trend.
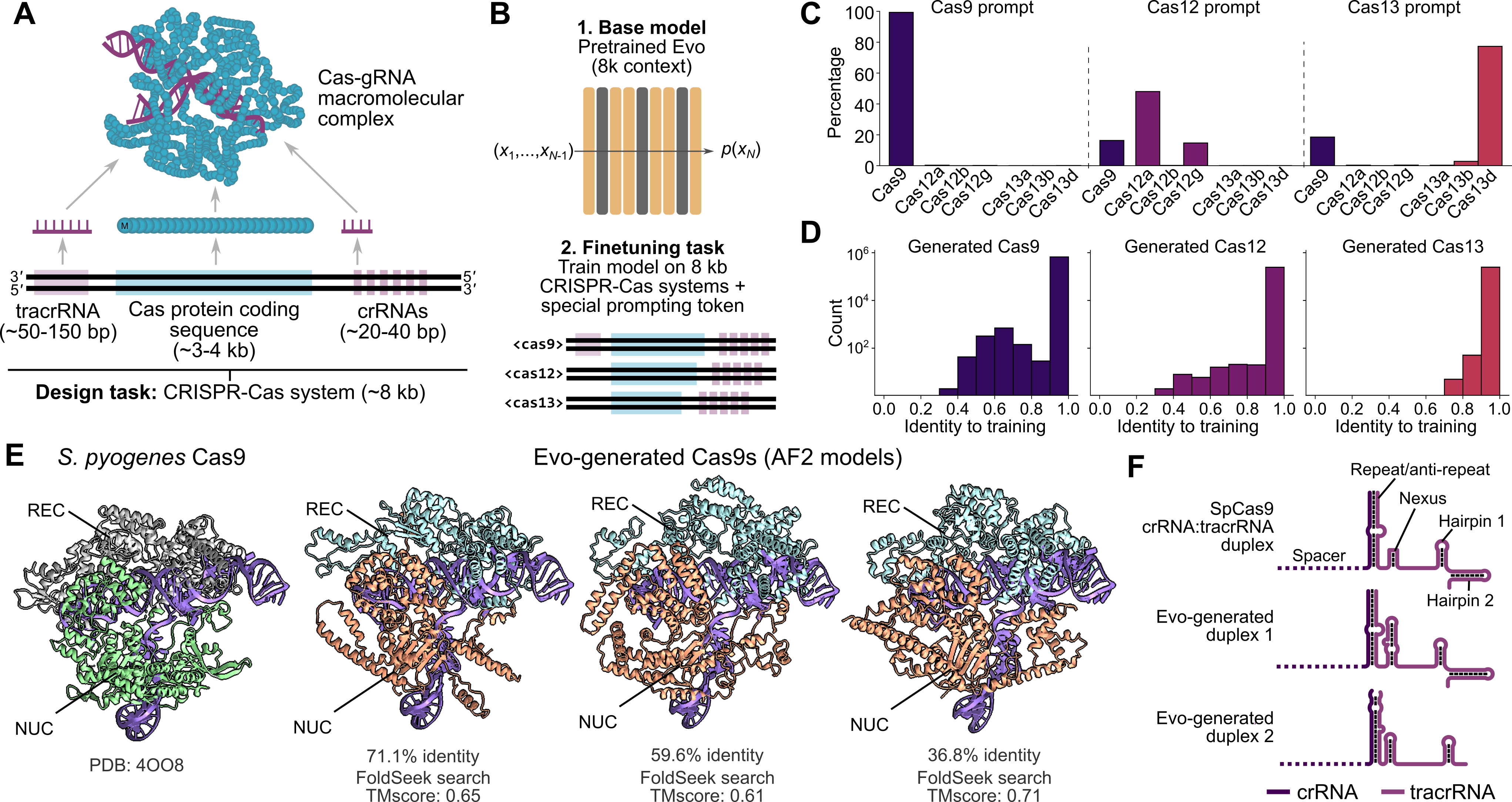 Figure 10: Evo CRISPR-Cas generation demonstrating de novo design of functional systems. Source: Nguyen et al., "Evo: Genomic Foundation Model", 202442
Figure 10: Evo CRISPR-Cas generation demonstrating de novo design of functional systems. Source: Nguyen et al., "Evo: Genomic Foundation Model", 202442
Similarly, Evo demonstrates that DNA foundation models can "generate coding-rich sequences up to 650 kb in length orders of magnitude longer than previous methods"52. This suggests future workflows will use fewer, more general models rather than task-specific architectures.
"Compared to HyenaDNA Evo is based on an improved hybrid design and scaled to 1000x larger model size and 100x more data"53. The architecture is "a hybrid of 29 layers of data-controlled convolutional operators interleaved with 3 layers (10%) of multi-head attention"54, combining the efficiency of convolutions with the expressiveness of attention where it matters most.
Conclusion
The genomic foundation model space offers genuine capability improvements, but the practical path forward requires matching model choice to your specific constraints. For most bioinformaticians, the recommendation is straightforward:
- Start small: DNABERT-2 or ESM-2 650M will handle most tasks with minimal infrastructure
- Use mean token pooling: It consistently outperforms alternatives
- Fine-tune with LoRA: Full fine-tuning is rarely necessary
- Validate on held-out data: Benchmarks don't transfer perfectly to your specific use case
The models exist. The weights are open. The practical barrier now is matching tools to problems. This guide should help you make that match faster.
The real story of these foundation models isn't just that they work. It's what it means for biology that they work at all. A neural network trained only to fill in blanks learned what we spent decades painstakingly cataloguing through experiment. The models don't make experimental structural biology obsolete. They need it for training data, and high-resolution applications still require experimental validation. But they've changed what's tractable.
Predicting the structure of every protein in a metagenome was fantasy five years ago. Now it's a two-week computation.
References
Footnotes
-
Lin, Z., et al. "ESM-2: Evolutionary Scale Modeling of Protein Sequences." Science, 2022. "more than 617 million structures" ↩ ↩2
-
Lin, Z., et al. "ESM-2: Evolutionary Scale Modeling of Protein Sequences." Science, 2022. "We were able to complete the predictions in 2 weeks on a cluster of approximately 2,000 GPUs" ↩
-
Dalla-Torre, H., et al. "The Nucleotide Transformer: Building and Evaluating Robust Foundation Models for Human Genomics." Nature Methods, 2024. ↩ ↩2 ↩3
-
Zhou, Z., et al. "DNABERT-2: Efficient Foundation Model and Benchmark for Multi-Species Genome." ICLR, 2024. "achieves comparable performance to the state-of-the-art model with 21x fewer parameters and approximately 92x less GPU time in pre-training" ↩
-
Zhou, Z., et al. "DNABERT-2: Efficient Foundation Model and Benchmark for Multi-Species Genome." ICLR, 2024. "DNABERT-2 117M parameters" ↩
-
Zhou, Z., et al. "DNABERT-2: Efficient Foundation Model and Benchmark for Multi-Species Genome." ICLR, 2024. "BPE significantly reduces the sequence length by approximately 5 times" ↩
-
Nguyen, E., et al. "HyenaDNA: Long-Range Genomic Sequence Modeling at Single Nucleotide Resolution." NeurIPS, 2023. "context lengths of up to 1 million tokens at the single nucleotide-level" ↩
-
Nguyen, E., et al. "HyenaDNA: Long-Range Genomic Sequence Modeling at Single Nucleotide Resolution." NeurIPS, 2023. "an up to 500x increase over previous dense attention-based models" ↩
-
Nguyen, E., et al. "HyenaDNA: Long-Range Genomic Sequence Modeling at Single Nucleotide Resolution." NeurIPS, 2023. "At sequence length 1M, HyenaDNA is 160x faster than its Transformer counterpart" ↩
-
Nguyen, E., et al. "HyenaDNA: Long-Range Genomic Sequence Modeling at Single Nucleotide Resolution." NeurIPS, 2023. "A Hyena operator can be evaluated in O(L log L) time" ↩
-
Dalla-Torre, H., et al. "The Nucleotide Transformer: Building and Evaluating Robust Foundation Models for Human Genomics." Nature Methods, 2024. "integrating information from 3202 diverse human genomes as well as 850 genomes from a wide range of species" ↩
-
Lin, Z., et al. "ESM-2: Evolutionary Scale Modeling of Protein Sequences." Science, 2022. "scale language models from 8 million parameters up to 15 billion parameters" ↩
-
Lin, Z., et al. "ESM-2: Evolutionary Scale Modeling of Protein Sequences." Science, 2022. "the largest language model of proteins to date" ↩
-
Lin, Z., et al. "ESM-2: Evolutionary Scale Modeling of Protein Sequences." Science, 2022. "prediction that is up to 60x faster than state-of-the-art while maintaining resolution and accuracy" ↩
-
Lin, Z., et al. "ESM-2: Evolutionary Scale Modeling of Protein Sequences." Science, 2022. "On a single NVIDIA V100 GPU, ESMFold makes a prediction on a protein with 384 residues in 14.2 seconds, 6x faster than a single AlphaFold2 model" ↩
-
Lin, Z., et al. "ESM-2: Evolutionary Scale Modeling of Protein Sequences." Science, 2022. "On shorter sequences the improvement increases up to approximately 60x" ↩
-
Lin, Z., et al. "ESM-2: Evolutionary Scale Modeling of Protein Sequences." Science, 2022. ↩
-
Cui, H., et al. "scGPT: Toward Building a Foundation Model for Single-Cell Multi-Omics." Nature Methods, 2024. "33 million scRNA-seq data of human cells" ↩ ↩2
-
Cui, H., et al. "scGPT: Toward Building a Foundation Model for Single-Cell Multi-Omics." Nature Methods, 2024. "51 organs/tissues and 441 studies" ↩
-
Theodoris, C., et al. "Transfer learning enables predictions in network biology." Nature, 2024. "larger pre-training data sizes yield superior pre-trained embeddings" ↩
-
Theodoris, C., et al. "Transfer learning enables predictions in network biology." Nature, 2024. "8 to 12% increase in the biological conservation score compared to the trained-from-scratch models" ↩
-
Zhou, Z., et al. "DNABERT-2: Efficient Foundation Model and Benchmark for Multi-Species Genome." ICLR, 2024. "About 14 days on 8 NVIDIA RTX 2080Ti V.S. 28 days on 128 NVIDIA A100" ↩ ↩2 ↩3
-
GenBench Authors. "GenBench: A Benchmarking Framework for Genomic Foundation Models." arXiv, 2024. "encompasses ten popular GFMs and performs extensive experiments across forty-three realistic datasets" ↩
-
GenBench Authors. "GenBench: A Benchmarking Framework for Genomic Foundation Models." arXiv, 2024. "Mean token embedding consistently and significantly improves sequence classification performance" ↩
-
GenBench Authors. "GenBench: A Benchmarking Framework for Genomic Foundation Models." arXiv, 2024. "four attention-based models and six convolution-based models" ↩
-
GenBench Authors. "GenBench: A Benchmarking Framework for Genomic Foundation Models." arXiv, 2024. "HyenaDNA and Caduceus utilize the hyena operator and state space model with complexity of O(L log L) and O(L) significantly lower than O(L^2) of attention-based models" ↩ ↩2
-
Nguyen, E., et al. "HyenaDNA: Long-Range Genomic Sequence Modeling at Single Nucleotide Resolution." NeurIPS, 2023. ↩ ↩2
-
GenBench Authors. "GenBench: A Benchmarking Framework for Genomic Foundation Models." arXiv, 2024. "the Coding Region comprising only about 1.5% of the genome is responsible for coding proteins" ↩
-
GenBench Authors. "GenBench: A Benchmarking Framework for Genomic Foundation Models." arXiv, 2024. "the vast Non-coding Region making up 98.5% of the genome plays crucial roles in gene regulation" ↩
-
Elnaggar, A., et al. "ProtTrans: Toward Understanding the Language of Life Through Self-Supervised Learning." IEEE TPAMI, 2021. "training on the Summit supercomputer using 5616 GPUs and TPU Pod up to 1024 cores" ↩
-
Dalla-Torre, H., et al. "The Nucleotide Transformer: Building and Evaluating Robust Foundation Models for Human Genomics." Nature Methods, 2024. "parameter-efficient fine-tuning technique requiring only 0.1% of the total model parameters" ↩ ↩2
-
Nguyen, E., et al. "HyenaDNA: Long-Range Genomic Sequence Modeling at Single Nucleotide Resolution." NeurIPS, 2023. "HyenaDNA effectively solves the task by using a context length of 450k to 1 million" ↩
-
Nguyen, E., et al. "HyenaDNA: Long-Range Genomic Sequence Modeling at Single Nucleotide Resolution." NeurIPS, 2023. "99.5% accuracy" ↩
-
Dalla-Torre, H., et al. "The Nucleotide Transformer: Building and Evaluating Robust Foundation Models for Human Genomics." Nature Methods, 2024. "the sequence representations alone match or outperform specialized methods on 12 of 18 prediction tasks" ↩
-
Dalla-Torre, H., et al. "The Nucleotide Transformer: Building and Evaluating Robust Foundation Models for Human Genomics." Nature Methods, 2024. "fine-tuned models matched or surpassed 15 of the 18 baselines with the largest and more diverse models constantly outperforming their smaller counterparts" ↩
-
Cui, H., et al. "scGPT: Toward Building a Foundation Model for Single-Cell Multi-Omics." Nature Methods, 2024. ↩
-
Cui, H., et al. "scGPT: Toward Building a Foundation Model for Single-Cell Multi-Omics." Nature Methods, 2024. "scGPT achieves an AvgBIO score of 0.821, which is 5-10% higher than the compared methods" ↩
-
Krishna, R., et al. "RoseTTAFold All-Atom." Science, 2024. "RFAA models 32% of cases successfully (< 2A ligand RMSD) compared to 8% for the AutoDock Vina server" ↩
-
Lin, Z., et al. "ESM-2: Evolutionary Scale Modeling of Protein Sequences." Science, 2022. "We fold over 617 million sequences from the MGnify90 database" ↩
-
Lin, Z., et al. "ESM-2: Evolutionary Scale Modeling of Protein Sequences." Science, 2022. "approximately 365 million predictions with good confidence" ↩
-
Lin, Z., et al. "ESM-2: Evolutionary Scale Modeling of Protein Sequences." Science, 2022. "more than 225 million high confidence predictions" ↩ ↩2
-
Nguyen, E., et al. "Evo: A 7-Billion Parameter Genomic Foundation Model." Science, 2024. ↩ ↩2 ↩3
-
Lin, Z., et al. "ESM-2: Evolutionary Scale Modeling of Protein Sequences." Science, 2022. "76.8% of high confidence predictions being separate from UniRef90 by at least 90% sequence identity" ↩
-
Lin, Z., et al. "ESM-2: Evolutionary Scale Modeling of Protein Sequences." Science, 2022. "12.6% without a match to experimentally determined structures" ↩
-
Nguyen, E., et al. "Evo: A 7-Billion Parameter Genomic Foundation Model." Science, 2024. "a 7 billion parameter genomic foundation model trained to generate DNA sequences at whole-genome scale" ↩
-
Nguyen, E., et al. "Evo: A 7-Billion Parameter Genomic Foundation Model." Science, 2024. "a context length of 131k tokens" ↩
-
Nguyen, E., et al. "Evo: A 7-Billion Parameter Genomic Foundation Model." Science, 2024. "OpenGenome dataset with over 80000 bacterial and archaeal genomes and millions of predicted prokaryotic phage and plasmid sequences covering 300B nucleotide tokens" ↩
-
Nguyen, E., et al. "HyenaDNA: Long-Range Genomic Sequence Modeling at Single Nucleotide Resolution." NeurIPS, 2023. "the human genome is 3.2B nucleotides" ↩
-
Elnaggar, A., et al. "ProtTrans: Toward Understanding the Language of Life Through Self-Supervised Learning." IEEE TPAMI, 2021. "ProtTrans models learned some of the grammar of the language of life" ↩
-
Gene Properties Language Models Study. arXiv, 2024. "we do not see a clear advantage for larger models, with cellPLM exhibiting comparable performance to the smaller ScGPT-H and even smaller Geneformer" ↩
-
Krishna, R., et al. "RoseTTAFold All-Atom." Science, 2024. "RoseTTAFold All-Atom (RFAA) a deep network capable of modeling full biological assemblies containing proteins nucleic acids small molecules metals and covalent modifications" ↩
-
Nguyen, E., et al. "Evo: A 7-Billion Parameter Genomic Foundation Model." Science, 2024. "Evo can generate coding-rich sequences up to 650 kb in length orders of magnitude longer than previous methods" ↩
-
Nguyen, E., et al. "Evo: A 7-Billion Parameter Genomic Foundation Model." Science, 2024. "Compared to HyenaDNA Evo is based on an improved hybrid design and scaled to 1000x larger model size and 100x more data" ↩
-
Nguyen, E., et al. "Evo: A 7-Billion Parameter Genomic Foundation Model." Science, 2024. "a hybrid of 29 layers of data-controlled convolutional operators interleaved with 3 layers (10%) of multi-head attention" ↩